酵素-2
あ
|
アーゼ (化学) -アーゼ(-ase)は、生化学において、酵素の命名に用いられる接尾辞である。酵素の最も一般的な命名では、基質名にこの接尾辞が付けられる。例えば、過酸化物(ペルオキシド)を分解する酵素はペルオキシダーゼ、テロメアを形成する酵素はテロメラーゼと呼ばれる。基質ではなく機能によって酵素を命名することもあり、例えばDNAを鎖状に重合(ポリメライズ)する酵素はポリメラーゼ、RNAから相補的DNAを逆転写する酵素はリバーストランスクリプターゼと呼ばれる。 この接尾辞は恐らく、ギリシア語のδιαστασις(分離)という言葉に由来するジアスターゼに由来している。 関連項目有機化合物のIUPAC命名法 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
C5050%阻害濃度(50%そがいのうど、IC50)または半数阻害濃度とは、化合物の生物学的または生化学的阻害作用の有効度を示す値である。どの濃度で、その薬物(毒など)が標的としている物の半数(50%)の働きを阻害できるかを示す。 目次概要IC50は元来英語の"half maximal (50%) inhibitory concentration"の略語であるが、現在ではIC50の表記の方が一般的である。しばしば対象にされる化合物とは医薬品候補化合物である。この定量的値は、特定の薬物もしくはその他の物質(阻害剤)が注目する生物学的プロセス(もしくはプロセスの要素、例えば酵素、細胞、受容体、微生物)の半数を阻害するにはどれだけの濃度が必要かを示し、より低い値を示す化合物は阻害剤としての活性がより高いと言える。IC50は薬学研究において阻害剤の有効性を示す値として広く用いられている。便宜上-log IC50で算出されるpIC50が代わりに用いられることもある。この場合より大きな値は指数関数的により大きな有効性を示している。FDA(アメリカ食品医薬品局)によればIC50はin vitroにおける50%阻害のために必要な薬物の濃度を表している[1]。 このことはEC50の場合と対照的である。EC50は50%の効果を示す濃度のことであるが、この場合はin vivoでの血漿中濃度を意味している[1]。 IC50の決定法機能的アンタゴニスト試験(Functional antagonist assay)ある薬物のIC50を決定するには、用量反応関係を明らかにし、異なった濃度のアンタゴニスト(薬物)がどのようにアゴニスト活性を抑制するかを調べる必要がある。注目するアンタゴニストのIC50値は、アゴニストの生物学的作用の最大値の半数が阻害されるために必要なアンタゴニストの濃度を決定することで算出される[2]。 IC50値は測定条件に依存する。一般的に、より高い濃度の阻害剤はアゴニスト活性をより低く抑える。IC50値は酵素の濃度が増加すると共に増加する。さらに、阻害の種類によりその他の因子がIC50値に影響することがある。ATP依存の酵素の場合、IC50値は(拮抗阻害の場合は特に)ATPの濃度と相互依存性を持つ。IC50値は2種類の阻害剤の有効性を比較するために用いられる。 競合結合試験この試験においては、単一濃度の放射性リガンド(通常アゴニスト)がそれぞれの試験管に使用される。通常、その解離定数値(Kd)と同じかより低濃度のリガンドが試験に用いられる。一定範囲の様々な濃度のその他の競合する非放射性化合物(通常拮抗剤)をそれぞれの試験管に加えた後、放射性リガンドの有効性を測定することで、特定の放射性リガンドの結合のレベルが決定される。競合曲線は計算によりロジスティック関数に回帰される。 この場合IC50は、放射性リガンドの全結合のうち50%が置換された時の競合するリガンドの濃度である。IC50値はチェン=プルソフ(Cheng-Prusoff)式に従い絶対阻害定数(Ki)に変換される[2][3]。 IC50と親和性チェン=プルソフ式によればIC50と親和性は少なくとも競争するアゴニストと拮抗剤の間では関連があると言えるが、IC50は親和性を示す直接的な指標ではない[4]。 ここで、Ki は阻害物質の結合親和性、 IC50は阻害物質の機能的強度、[S]は基質の濃度、Kmは酵素活性が最大値の半分となる時の基質の濃度である(Kmはしばしば基質の酵素に対する親和性と混同されるが誤りである)。 その他に、細胞受容体に対する阻害定数については以下の式で表わされる[5]。 ここで [A] はアゴニストの固定濃度、EC50は受容体の最大活性の半分を生じさせるアゴニストの濃度である。ある化合物のIC50値は放射性リガンドの濃度に依存して実験毎に変化しうるが、Kiは絶対的な値である。Kiは薬物の阻害定数であり、放射性リガンドが存在しないならば、競合試験において受容体の50%を占める競合リガンドの濃度となる[3]。 チェン=プルソフ式は高いアゴニスト濃度においてよい推定値を与えるが、低いアゴニスト濃度では、Kiを実際より高くあるいは低く見積もる。これらの条件では、その他の解析法が推奨されている[5]。 脚注1. ^ a b アメリカ食品医薬品局. “IC50 versus EC50”. 2011年8月23日閲覧。 2. ^ a b NIH Chemical Genomics Center (2008年). “Assay Guidance // Assay Guidance Manual // Assay Operations for SAR Support”. 2011年8月23日閲覧。 3. ^ a b Glaxo Wellcome and Science - Global. “Pharmacology Guide - Receptor binding techniques: competition (inhibition or displacement) assays”. 2011年8月23日閲覧。 4. ^ Cheng Y, Prusoff WH (December 1973). “Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction”. Biochem. Pharmacol. 22 (23): 3099–108. PMID 4202581. 5. ^ a b Lazareno S, Birdsall NJ. (1993). “Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations”. Br. J. Pharmacol. 109 (4): 1110-1109. PMC 2175764. PMID 840192. 関連項目外部リンク |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アイソザイム アイソザイム(Isozyme;アイソエンザイム Isoenzymeともいう)とは、酵素としての活性がほぼ同じでありながら、タンパク質分子としては別種である(アミノ酸配列が異なる)ような酵素をいう。 解説 アイソザイムについて厳密には、全く別の遺伝子に由来する狭義のアイソザイムと、同じ種類の遺伝子(ただし別の個体の遺伝子、または同一個体中の対立遺伝子であって、配列がわずかに異なる)に由来するアロザイム(Allozyme)に分けられるが、いずれもまとめてアイソザイムと呼ぶことが多い。 狭義のアイソザイムには、個体の発達に伴って比率が変化する(例えば乳児と成人とで種類が異なる)ものもある。 また血液中の酵素には疾患によってアイソザイムの比率が変化するもの(代表的なものとして逸脱酵素の乳酸脱水素酵素(LDH)など)もあり、アイソザイムの分析は疾患の種類や部位を特定する上で重要である。 アイソザイム分析法としては、酵素阻害剤による活性の変化、分子量や等電点(電気泳動を用いる)、抗原抗体反応によるものなどが用いられる。 アイソザイムは遺伝子型を反映しているので、間接的な“遺伝子マーカー”として利用できる。そのためアイソザイム分析は、1960年代以降、生物の分類や、個体・個人の遺伝的性質に関する研究などに盛んに用いられた。また多数の個体をまとめて電気泳動にかけて分析することで遺伝子頻度の計算が比較的容易に可能であるため、集団遺伝学のツールとしても盛んに用いられている。現在では、より直接的に目的の遺伝子DNAまたは遺伝子マーカーを調べる方法(分子分類学、DNA鑑定など)にとって代わられつつある。 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
β-ラクタマーゼ
β-ラクタマーゼ(ベータラクタマーゼ、β-lactamase)とはβ‐ラクタム系抗生物質を加水分解する酵素である。ペニシリン/セファロスポリンアミド-β-ラクタムヒドロラーゼ (penicillin/cepharosporin amido-β-lactam hydrolase)とも呼ばれる。EC3.5.2.6に分類される酵素である。 幾つかの種類のグラム陰性菌がβ-ラクタマーゼを産生することでβ-ラクタムに対して耐性を示すことが知られている。なお、β-ラクタム耐性はβ-ラクタマーゼのみが原因ではなくMRSAのようにペニシリン結合タンパク質の基質特異性が変化しても現れる。 現在β-ラクタマーゼは基質特異性の違いにより · ペニシリナーゼ (クラスA β-ラクタマーゼ) · メタロ-β-ラクタマーゼ (クラスB β-ラクタマーゼ、亜鉛-β-ラクタマーゼ、カルバペネマーゼ) · セファロスポリナーゼ (クラスC β-ラクタマーゼ) · オキサシリナーゼ (クラスD β-ラクタマーゼ) これら4種のβ-ラクタマーゼのうち、クラスB β-ラクタマーゼは活性中心に亜鉛を持つが、他はセリン残基を持つ。ペニシリナーゼはペニシリン系抗生物質と第二世代セファロスポリンを分解するのに対して、セファロスポリナーゼは主にセファロスポリンを分解する。オキサシリナーゼはオキサシリンをも分解するペニシリナーゼであり、メタロ-β-ラクタマーゼはカルバペネム系抗生物質を分解する点に特徴がある。 β-ラクタマーゼの遺伝子は、細菌の染色体上あるいはプラスミド上に存在する。特に伝達性薬剤耐性プラスミド (drug resistance plasmid)に存在するβ-ラクタマーゼ遺伝子は菌種特異性も少なく多剤耐性菌の発生にも関与していると考えられる。 脚注1. ^ β-ラクタム耐性菌とその検出方法、関東化学 2. ^ Bush, K. et. al. A functional classification scheme for β-lactamases and its correlation with molecular structure, Antimicrob Agents Chemother., 39, 1211-1233, 1995. 3. ^ Ambler, R. P., The structure of β-lactameses, Philos Trans R Society Lond (Biol), 289, 321-331, 1980. 4. ^ 石井良和、基質特異性拡張型β-ラクタマーゼ産生大腸菌、クレブシエラ、臨床と微生物、26、121-125, 1999. 関連項目クラブラン酸· 薬剤耐性 · ペニシリン · セファロスポリン 出典· β-ラクタマーゼ『生物学辞典』第4版、岩波書店。 · β-ラクタマーゼについて 日本ベクトン・ディッキンソン株式会社 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アスパラギナーゼ
アスパラギナーゼ(英: asparaginase)(正確にはL-アスパラギナーゼ)とはアスパラギンのアスパラギン酸への加水分解を触媒する酵素の一つ。アスパラギナーゼは急性リンパ性白血病の治療用に協和発酵キリンからロイナーゼ(Leunase)の商品名で市販されており、肥満細胞腫のプロトコールにも使用される[1]。他の化学療法剤と異なり、組織障害の危険性がなく、筋肉内、皮下、静脈内投与が可能である。 概要 アスパラギナーゼは血中のL-アスパラギンを分解し、アスパラギン要求性の腫瘍細胞を栄養欠乏にすることにより抗腫瘍効果を発揮するとされる。現在日本で承認されている唯一のアスパラギナーゼである「ロイナーゼ」は大腸菌(Escherichia coli)由来のものである[2]。米国などの海外ではErwinia chrysanthemi由来のアスパラギナーゼが使用できる。大腸菌由来とErwinia chrysanthemi由来の治療成績を比較すると、大腸菌由来の方が治療成績が良いがアレルギーを示す患者もあり、これに対してはErwinia chrysanthemi由来のアスパラギナーゼが代替薬となりえる[3]。Erwinia chrysanthemi由来のアスパラギナーゼである「エルウィニア」は学会からの要望もあり、2010年に大原薬品工業が開発要請を受けて[4]、現在治験が進行中である[5]。 出典 ^ Appel IM, van Kessel-Bakvis C, Stigter R, Pieters R (2007). “Influence of two different regimens of concomitant treatment with asparaginase and dexamethasone on hemostasis in childhood acute lymphoblastic leukemia”. Leukemia 21: 2377. doi:10.1038/sj.leu.2404793. PMID 17554375. ^ Kyowa Hakko Kirin "LEUNASE" ^ Michel Duval, Stefan Suciu, Alina Ferster, Xavier Rialland, Brigitte Nelken, Patrick Lutz, Yves Benoit, Alain Robert, Anne-Marie Manel, Etienne Vilmer, Jacques Otten, and Noël Philippe (2002). “Comparison of Escherichia coli–asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer—Children’s Leukemia Group phase 3 trial”. Blood 99 (8): 2734-2739. doi:10.1182/blood.V99.8.2734. PMID 11929760. ^ 大原薬品工業 厚生労働省より「エルウィニア L-アスパラギナーゼ」の開発要請を受ける-2011年1月31日 ^ 白血病治療剤「エルウィニア L-アスパラギナーゼ」の治験開始について-2011年12月7日 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
アスパラギンシンテターゼアスパラギンシンテターゼ (asparagine synthetase、アスパラギン合成酵素; EC 6.3.1.1) はL-アスパラギン酸とアンモニアからL-アスパラギンを生合成する酵素。アスパラギン酸‐アンモニアリガーゼ (aspartate-ammonia ligase) とも呼ばれる。 アデノシン三リン酸を1分子消費し、アデノシン一リン酸とピロリン酸を生成する反応を可逆的に触媒する。 ATP + L-aspartate + NH3 = AMP + diphosphate + L-asparagine この酵素は古細菌、真正細菌、真核生物に広く存在している。ヒトの培養細胞を用いた研究ではアミノ酸が欠乏すると転写量が上がることが知られている。 出典[
外部リンクIUBMB entry for 6.3.1.1(英語)関連項目· リガーゼ |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アスパラギンシンテターゼ アスパラギンシンテターゼ (asparagine synthetase、アスパラギン合成酵素; EC 6.3.1.1) はL-アスパラギン酸とアンモニアからL-アスパラギンを生合成する酵素。アスパラギン酸‐アンモニアリガーゼ (aspartate-ammonia ligase) とも呼ばれる。 アデノシン三リン酸を1分子消費し、アデノシン一リン酸とピロリン酸を生成する反応を可逆的に触媒する。 ATP + L-aspartate + NH3 = AMP + diphosphate + L-asparagine この酵素は古細菌、真正細菌、真核生物に広く存在している。ヒトの培養細胞を用いた研究ではアミノ酸が欠乏すると転写量が上がることが知られている。 出典
外部リンク
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
アルデヒドデヒドロゲナーゼアルデヒドデヒドロゲナーゼ (aldehyde dehydrogenase、ALDH) はアルデヒドを酸化してカルボン酸にする反応を触媒する酵素である。 生物に普遍的に存在し、ヒトには17種類のALDHファミリータンパク質が存在する[1]。 ヒトのALDHの例ALDH1A (RALDH) レチナールの酸化によりレチノイン酸を作り出す酵素。レチノイン酸はビタミンAが生体内で働く際の本体で、目や骨の形成など様々な分化過程に関わる。このため、ALDH1A の機能に異常があると正常に発生が進行しない。分子量は約 55 kDa。四量体として機能する。ALDH1A1 (RALDH1)、ALDH1A2 (RALDH2)、ALDH1A3 (RALDH3) の3種があり、それぞれ異なった組織発現様式を示す。 ALDH2 (ALDH I) 肝臓、心臓、腎臓、筋肉に多く存在する。細胞内ではミトコンドリアに局在するがミトコンドリアDNAにコードされるミトコンドリア遺伝子ではなく核ゲノム遺伝子に由来する。一般にアルコールに弱い人はアルコールに強い人に比べて持っている ALDH2 の活性が弱い。 ALDH2 遺伝子には少なくとも4種の対立遺伝子が報告されているが、日本人が一般に持つのは ALDH2*1 と ALDH2*2 で、ALDH2*2 が機能喪失型。四量体として機能し、ALDH2*2 を持つ複合体は機能を持たないため、ヘテロ接合型でも ALDH2 の活性が極端に下がる。 ALDH9A1 γ-アミノブチルアルデヒドから神経伝達物質であるγアミノ酪酸 (GABA) を作る。494 アミノ酸、分子量 54 kDa。四量体として機能する。 肝臓、副腎、腎臓で高い酵素活性が認められた一方で、ノーザンブロットでは筋肉で最も高いmRNAの存在が確認された[2] 。同様にノーザンブロットにより、脳の中では脊髄で最も高い発現がみられた[3]。 脚注1. ^ Sladek, N. E. (2003). "Human aldehyde dehydrogenases: potential pathological, pharmacological, and toxicological impact". J. Biochem. Mol. Toxicol. 17: 7–23. 2. ^ Izaguirre, G.; Kikonyogo, A.; Pietruszko, R. (1997). "Tissue distribution of human aldehyde dehydrogenase E3 (ALDH9): comparison of enzyme activity with E3 protein and mRNA distribution". Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 118: 59–64. 3. ^ Kikonyogo, A.; Pietruszko, R. (1996). "Aldehyde dehydrogenase from adult human brain that dehydrogenates gamma-aminobutyraldehyde: purification, characterization, cloning and distribution". Biochem. J. 316: 317–324. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
光活性化アデニル酸シクラーゼ 光活性化アデニル酸シクラーゼ(ひかりかっせいかアデニルさんシクラーゼ、photoactivated adenylyl cyclase; PAC)とはミドリムシ(Euglena gracilis)から発見された光センサータンパク質。Iseki らにより2002年にイギリスの科学雑誌 Nature 誌上で発表された[1]。通称名はその頭文字をとって PAC(パック)と呼称される。このタンパク質は酵素としてはアデニル酸シクラーゼであるが、光を感知することで cAMP を作ることからこの名が付けられた。アデニル酸シクラーゼは生物界に広く分布する酵素であるが、光によって活性が調節されるアデニル酸シクラーゼは極めて珍しいため注目された。 目次 [非 細胞内での局在 ミドリムシの鞭毛(長鞭毛)の付け根近くには小さな膨らみが存在し、これを鞭毛膨潤部(paraflagellar body; PFB)と呼ぶ。この膨らみを構成する主要成分が PAC である。鞭毛膨潤部は蛍光顕微鏡下(UV励起または青色励起)で緑色の蛍光を発するが、これは PAC に結合していたフラビン色素(FAD)によるものと考えられている。鞭毛膨潤部こそミドリムシの真の目であるが、これは眼点と呼ばれるカロテノイドでできた偽の目に取り囲まれている。この見かけ上の眼点は鞭毛膨潤部に差し込む光の一部を遮蔽することで、鞭毛膨潤部の光感知能に指向性を持たせる役割を果たすと考えられている。 分子の構造と機能 PAC は FAD を結合する光感知領域と、cAMP を合成する酵素領域からなるタンパク質から成る。PAC には2種類の分子があり、それぞれ PACα、PACβ と呼ばれている。分子量は前者が 105kDa、後者が 90kD であるが、いずれの分子にも光感知領域と酵素領域が2ヶ所ずつ存在する。生体内ではこれらがヘテロ4量体を形成していると推定されている。フラビン色素を結合する光感知領域の一次構造は BLUF(sensor protein of Blue Light Using FAD)と呼ばれる一連のフラビンタンパク質と相同性が認められている。 生物界での分布 PAC は葉緑体を有するミドリムシ類からのみ報告されている。 注釈・参考文献 ^ Iseki M, Matsunaga S, Murakami A, Ohno K, Shiga K, Yoshida K, Sugai M, Takahashi T, Hori T, Watanabe M (2002). “A blue-light activated adenylyl cyclase mediates photoavoidance in Euglena gracilis”. Nature 415 (6875): 1047-51. PMID 11875575 関連項目 ユーグレナ藻 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ATPアーゼATPアーゼ(ATPエース、ATPase、ATPases (ion transport))とは、アデノシン三リン酸 (ATP) の末端高エネルギーリン酸結合を加水分解する酵素群の総称である(EC番号 3.6.1.3、3.6.3、3.6.4)。ATP は生体内のエネルギー通貨であるから、エネルギーを要する生物活動に関連したタンパク質であれば、この酵素の活性を持っていることが多い。 日本語ではATPアーゼを「アデノシン三リン酸分解酵素」などと表現できる。なお、「ホスファターゼ」は「リン酸分解酵素」のことであるから、「アデノシン三リン酸ホスファターゼ」という呼び方は「リン酸」の重言となり、正しくない。 特徴ATPアーゼは以下の反応を触媒する酵素の総称である。 この時に発生するエネルギーを利用して、エネルギーを要する生物体内作用に寄与している。通常は ATP 以外のヌクレオチド三リン酸(GTP、UTP、CTPなど)に作用することが知られている。しかしながら存在している部位によって少しずつ性状が異なっている。 ATPに共通する特性として、スルフヒドリル基(SH基)を必要とすることと、Mg2+, Ca2+ によって活性化あるいは阻害を受けるという点が挙げられる。 役割ATPアーゼの役割はエネルギーの関与する全ての反応に寄与していると言ってよい。
種類運動性タンパク質ATPアーゼミオシンアクチン系に代表されるATPアーゼである。ATP加水分解によるコンフォメーションの変化を受けることを特徴とする。タンパク質にATPが結合することによってタンパク質の立体構造に変化が起こり、その構造変化を利用して実際にタンパク質(ひいては細胞を)を「稼動」させることに関係している。 ミオシン、ダイニン、キネシンはそれぞれが蛍光標識を用いた一分子観測でその稼動が観察されている。
イオン輸送性ATPアーゼATPの加水分解エネルギーを使って生体膜を透過しないイオンの輸送を行うATPアーゼの一群である。ATP合成酵素もこれに分類される。F型、A型、V型、P型が存在している。P型をのぞくものは構造がよく似ており、イオン(主にプロトン)駆動型モーター (Fo, Ao, Vo) ならびにATP駆動型モーター (F1, A1, V1) から形成される。 全てがイオン濃度勾配を用いてATP合成および逆反応のATP加水分解に伴うイオン濃度勾配の形成が可能である。しかしながら、ATP合成酵素として用いられているのはF型およびA型のみである。
ABC ATPアーゼABC とは ATP Binding Cassette (ATP結合カセット)の略称である。細胞への物質取り込みおよび排出に関係する。膜貫通型の ABC ATPアーゼは、常に4つの機能ドメイン(2つの膜貫通ドメインと2つのABCドメイン)から構成される。これらのドメインは全てが一つの遺伝子にコードされている場合もあれば、それぞれ別々の遺伝子にコードされている場合もある。膜貫通ドメインの配列は多様であるが、ABCドメインと呼ばれるATP結合部位の配列は高度に保存されている。真核生物(主にヒト)では有害物質の排出に使用されているが、一方原核生物では糖、アミノ酸と言った物質の取り込みに用いられている。また、ヒトの中でもトランスポーター、チャネル、レセプター等、その機能は多彩である。 これら生体膜貫通型の古典的なABC ATPアーゼに加え、最近ではDNA結合型の ABC ATPアーゼが知られるようになってきている。代表的なものとして、染色体の高次構造と機能を制御するSMCタンパク質があり、これらはコンデンシンあるいはコヒーシンのコアサブユニットとして機能する。また、DNA2重鎖切断の修復に関与する Rad50 もこのカテゴリーに属する。 ABCドメインの特徴は、多くのATPアーゼが共有する Walker A と Walker B モチーフに加え、Signature モチーフ(あるいはCモチーフ)と呼ばれる配列を持つことにある。すべての ABC ATPアーゼは一対のABCドメインをもち、2つのATP分子は2つのドメインに挟まれるようにして結合する。この際、ATPは一方のドメインの Walker A と Walker B モチーフに結合し、もう一方のドメインのCモチーフと接触する。このCモチーフとの接触が、ATPの加水分解に必須である。すなわち、ATPの結合と加水分解のサイクルが2つのABCドメインの会合と解離のサイクルを制御し、さらにその構造変換が基質結合ドメイン(例えば、ABCトランスポーターの膜貫通ドメイン)に伝達されると考えられている。その作用メカニズムは、2ストロークエンジンに例えられることもある。
AAA ATPアーゼAAA とは ATPases Associated with diverse cellular Activities の略称である。タンパク質の細胞内小器官への輸送(プロテインキネシス)、膜融合、細胞内小器官の形成、DNA複製、転写調節など機能は多様だが、全てがリング状オリゴマー構造を取っている。ATPの加水分解エネルギーはタンパク質のアンフォールディング(3次構造をほどく)やタンパク質分解、オリゴマーの拡大などに使用されていると考えられている。 真核細胞のみならず、細菌(大腸菌)、古細菌からも見つかっている。 課題運動性タンパク質ATPアーゼを除く全てのタンパク質が生体膜に存在している。そのため構造が理解されていないことが多く、未開拓な酵素の一つである。また、ATPアーゼ活性そのものについてもよくわかっておらず、ATPのエネルギーを得た中間体などの解析から「エネルギーを持ったタンパク質」の状態を理解することへの研究もなされている。 最も研究が進んでいるであろうATPアーゼはミオシンおよびATP合成酵素であるが、その全てが理解されたとはいずれも考えられない状況であることは否めない。 関連項目 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アデノシンデアミナーゼ この記事には複数の問題があります。改善やノートページでの議論にご協力ください。 出典がまったく示されていないか不十分です。内容に関する文献や情報源が必要です。(2015年4月) 正確性に疑問が呈されています。(2015年4月) リボンモデル アデノシンデアミナーゼ(ADA:adenosine deaminase; EC 3.5.4.4)は、細胞内で核酸の代謝に関わる酵素である。これが先天的に欠損していると重篤な免疫不全の原因になる。また、結核の診断において胸水・髄液中ADA活性の上昇が特徴的として知られており、臨床的に利用されている。 働きは、核酸塩基の一種アデノシンを分解しイノシンとアンモニアを生成することである。同様にアデノシンを代謝するものとしてアデノシンキナーゼが存在するが、ADAはアデノシン濃度が高いときに特に働いている。 血液腫瘍(白血病など)、肝炎などで高値を示すほか、胸水が結核性の場合は細菌性・心原性のときに比べて胸水中ADA濃度が上昇する。髄膜炎でも同じく、結核性髄膜炎では髄液中ADA濃度が高値を示す。 ADA欠損症(重症複合免疫不全症) ADAはリンパ球増殖の際に特に需要が高く活性が上がるが、生まれつきADAを合成できない場合はリンパ球が減少するなどして免疫不全を来す。無治療の場合は多くが乳児期に死亡するという重篤な疾患である。 治療としては、ADA酵素を外部から補充する治療が一般的である。 しかしこれは、ADAを合成する遺伝子の欠損であるため究極的な治療法は遺伝子治療であり、最も早くから遺伝子治療の研究対象となってきた。 手法としてはウイルスをベクター(運び屋)として患者のリンパ球にADA合成遺伝子を組み込み、体内に戻すというものである。 出典 IUBMB entry for 3.5.4.4(英語) BRENDA references for 3.5.4.4 (英語) PubMed references for 3.5.4.4(英語) PubMed Central references for 3.5.4.4(英語) Google Scholar references for 3.5.4.4(英語) 外部リンク[編集] IUBMB entry for 3.5.4.4(英語) KEGG entry for 3.5.4.4(英語) BRENDA entry for 3.5.4.4(英語) NiceZyme view of 3.5.4.4(英語) EC2PDB: PDB structures for 3.5.4.4(英語) PRIAM entry for 3.5.4.4(英語) PUMA2 entry for 3.5.4.4(英語) IntEnz: Integrated Enzyme entry for 3.5.4.4(英語) MetaCyc entry for 3.5.4.4(英語) Atomic-resolution structures of enzymes belonging to this class(英語) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アナログ耐性 アナログ耐性(アナログたいせい)とは、主に生物の変異株が持つ、フィードバック阻害を司る代謝系の最終生産物に類似の物質(アナログ)の存在下でも生存できる性質。アナログの存在下で生物を培養した場合、通常は代謝系の酵素反応の一部がアナログによってフィードバック阻害を受けるため、生存に必要な物質を生産できずに死滅してしまうが、アナログ耐性を獲得した変異株はアナログによるフィードバック阻害を受けないため生存が可能であり、さらに通常よりも多くの物質を生産できる。このようにして得られた変異株はアナログ耐性株と呼ばれ、インスリン等の薬剤の大量合成や、うまみ成分の多い稲・トウモロコシ等の作物の品種改良、アミノ酸のL-リシンを蓄積した遺伝子組換えトウモロコシ(「遺伝子組み換え作物」参照)、香気成分の多い清酒酵母の開発などに利用されている。特に成功した例としては、微生物を用いたアミノ酸発酵や核酸発酵である。 アミノ酸発酵への応用 アミノ酸のL-リシンとL-トリプトファンとL-スレオニンのアナログ耐性株によるアミノ酸発酵について解説する。 生合成されたリシンによるフィードバック阻害は、リシン生合成系の酵素群の中の鍵酵素でもあるジヒドロジピコリン酸合成酵素(dihydrodipicolinate synthase: DHDPS, EC 4.2.1.52, 反応)やアスパラギン酸リン酸化酵素(aspartate kinase, EC 2.7.2.4, 反応)の活性をリシンが阻害することに依存している。一方、リシンのアナログであるS-(2-アミノエチル)-L-システイン(CAS No: 4099-35-8)耐性株であるリシン生産菌は、リシンとスレオニンによるアスパラギン酸リン酸化酵素に対する協奏的なフィードバック阻害(リシンとスレオニンが同時に存在すると阻害されるが、各々単独に存在する場合は阻害されない)が解除されている。そのため、生合成されたリシンによってリシン生合成系が阻害されず、リシンやスレオニンが同時に高濃度で存在していてもリシン生合成が続き、大量のリシンが生合成される。DHDPSの変異によるリシンのフィードバック阻害が解除された株でも生合成されたリシンによってリシン生合成系が阻害されず、大量のリシンを培地中に放出する。 トリプトファンのアナログである5-メチルトリプトファン(CAS No: 951-55-3)や5-フルオロトリプトファン(CAS No: 16626-02-1)耐性株では、アンスラニル酸合成酵素(anthranilate synthase, EC 4.1.3.27, 反応)に対するトリプトファンによるフィードバック阻害が解除されており、トリプトファンを蓄積できる。 スレオニンのアナログである2-アミノ-3-ヒドロキシ吉草酸耐性変異株では、ホモセリン脱水素酵素(homoserine dehydrogenase, EC 1.1.1.3, 反応)に対するスレオニンによるフィードバック阻害が解除されており、スレオニンを蓄積できる。 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ホロ酵素(アポ酵素から転送) ホロ酵素(ホロこうそ、英: holoenzyme)とは、酵素本体となるタンパク質分子に、非タンパク質性の分子が結合して初めて酵素として機能するものを呼ぶ。この場合の非タンパク質性の分子の部分を補因子と呼ぶ。補酵素を要求する酵素はホロ酵素であり、補酵素部分が補因子となっている。 多くの場合、非タンパク質性の部分を失うと活性を失う。このタンパク質部分のみの状態のものをアポ酵素と呼ぶ。 また、複数のタンパク質分子が複合体を形成して初めて活性を示すような酵素についても、ホロ酵素と呼ぶ場合がある。この場合、一部のサブユニットを失って活性を失った状態のものがアポ酵素と呼ばれる。 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アポリポタンパク質 この記事は検証可能な参考文献や出典が全く示されていないか、不十分です。 出典を追加して記事の信頼性向上にご協力ください。(2014年2月) アポリポタンパク質(アポリポタンパクしつ、英語: Apolipoprotein)は、リポタンパク質と結合し、リポタンパク質の認識や脂質代謝に関与する酵素群の活性化あるいは補酵素として働く一群のタンパク質である。アポ(apo-)はギリシア語系の接頭語で「~を切り離した、~をまぬがれる」という意。またリポ(lipo-)は脂質の意。 種類 アポリポタンパク質は、構造やはたらきによりアポリポタンパク質AからEまでの5種に大別される。さらに、それらのいくつかは、アポリポプロテインA-IやC-IIのようにサブクラスに分けられる。 アポリポプロテインA-I(apo A-I) apo A-IはHDLの主要な構成成分であり、HDLの代謝に関与している。apo A-I遺伝子を欠損したマウスでは血中の高密度リポタンパク質(英語版)(HDL)濃度が著しく減少することが知られている。 アポリポプロテインA-II(apo A-II) apo A-IIはHDLの二番目に主要な構成成分であり、HDLの代謝に関与している。マウスにおける老化アミロイドーシスの原因タンパク質としてアミロイド繊維を作る。 アポリポプロテインB-48(apo B-48) 粗面小胞体で合成された後、ゴルジ体へと輸送される過程で、糖鎖が付加されて成熟する。apo B-48の名前は、apo B遺伝子でコードされるタンパク質の内、N末端側の48%で構成されていることに由来する。これは、小腸においてapo Bが合成される際、mRNAが転写後に、その核酸塩基がシトシンからウラシルへと変換され、途中に終止コドンが生成するためである。apo B-48は合成後、カイロミクロンに組み込まれて、小腸からの脂質吸収に必須な役割を果たす。 アポリポプロテインB-100(apo B-100) apo B-100はapo B遺伝子にコードされるタンパク質で、4,536アミノ酸残基よりなる非常に大きな分子である。apo B100は肝臓で合成され、超低密度リポタンパク質(英語版) (VLDL)の構成成分となる。他のアポリポタンパク質と異なり、VLDLとHDLとの間で相互に受け渡しが行われない。apo B-100はVLDLおよび低密度リポタンパク質(英語版) (LDL)に存在し、LDL受容体の主要なリガンドとして働く。 アポリポプロテインC-II(apo C-II) apo C-IIはリポタンパク質が細胞に脂質を受け渡す際に必要な酵素であるリポプロテインリパーゼを活性化するのに必要となる分子である。apo C-IIはカイロミクロン、VLDLが成熟する際に、HDLから受け渡され、それらが末梢への脂質輸送を終えたときHDLに戻される。 アポリポプロテインE(apo E) apo C-IIと同様に、カイロミクロン、VLDLおよびLDLとHDL間で受け渡しと再利用が行われるタンパク質である。apo Eの役割は、細胞にこれらのリポタンパク質が認識されるときのマーカーとなる事である。すなわち、肝臓などにおけるLDL受容体に代表されるリポ脂質に対する受容体のリガンドとなる。 apo EにはE2、E3およびE4の3種の分子種が知られている。それぞれの相違はアミノ酸配列の112番目と158番目にあり、「正常型(野生型)」といわれるapo E3では112番目がシステイン、158番目がアルギニンとなっているが、apo E4では112番目がアルギニン、apo E2では158番目がシステインとなっている。 Apo E2は受容体との親和性が弱く、家族性III型脂質異常症の原因因子である。また、apo E4はアルツハイマー病の危険因子として知られており、現在因果関係が活発に研究されている。アメリカでの調査では、apo Eの遺伝子型ε2、ε3およびε4の出現頻度はそれぞれ8%、78%および14%である。なお、他にapo E1、E5およびE7の存在が報告されているが、出現頻度は極めてまれである。 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
β-グルコシダーゼβ-グルコシダーゼ(β-glucosidase; EC 3.2.1.21)は糖のβ-グリコシド結合を加水分解する反応を触媒する酵素。β‐D‐グルコシドグルコヒドロラーゼ,アミグダーゼとも呼ばれる[1]。また、β-グリコシド結合を持つ代表的な糖であるセロビオースやゲンチオビオースから、しばしばセロビアーゼ、ゲンチオビアーゼとも呼ばれる。 微生物,高等植物,動物の肝臓・腎臓・小腸粘膜,カタツムリ消化液などに広く分布するが、基質特異性は起源によって異なる[1]。 α-グルコシダーゼ同様、動植物通じて広く存在し、異化代謝に関わっている。アグリコンと糖の結合も分解するが、アグリコンの構造によっては、基質が阻害剤となる場合もある。セルロースの分解に関連する酵素で、β-グルコシダーゼの活性が低いとセロビオースが蓄積し、セルロースの働きを阻害する場合がある。ただし、一般的にはセルラーゼの活性の方が低い。 β-グルコシダーゼの先天性欠損症はゴーシェ病を引き起こす[1]。 出典1. ^ a b c β-グルコシダーゼ、『生物学辞典』、第4版、岩波書店 · IUBMB entry for 3.2.1.21(英語) · BRENDA references for 3.2.1.21 (英語) · PubMed references for 3.2.1.21(英語) · PubMed Central references for 3.2.1.21(英語) · Google Scholar references for 3.2.1.21(英語) 外部リンク· IUBMB entry for 3.2.1.21(英語) · KEGG entry for 3.2.1.21(英語) · BRENDA entry for 3.2.1.21(英語) · NiceZyme view of 3.2.1.21(英語) · EC2PDB: PDB structures for 3.2.1.21(英語) · PRIAM entry for 3.2.1.21(英語) · PUMA2 entry for 3.2.1.21(英語) · IntEnz: Integrated Enzyme entry for 3.2.1.21(英語) · MetaCyc entry for 3.2.1.21(英語) · Atomic-resolution structures of enzymes belonging to this class(英語) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
アミダーゼ アミダーゼ(Amidase、EC 3.5.1.4)は、次の反応を触媒する酵素である。 モノカルボン酸アミド+H2O ⇌ {\displaystyle \rightleftharpoons } モノカルボン酸+NH3 つまり、この酵素の基質はモノカルボン酸アミドと水の2種で、生成物はモノカルボン酸とアンモニアである。 この酵素は加水分解酵素の1つで、ペプチド結合とは別の炭素-窒素結合、特に直線的なアミドに作用する。 この酵素の組織名は、アシルアミドアミドヒドロラーゼ(acylamide amidohydrolase)である。別名に、アシルアミダーゼ(Acylamidase)やアシラーゼ(Acylase)、アミドヒドロラーゼ(amidohydrolase)、デアミナーゼ(deaminase)、fatty acylamidase、そして、N-アセチルアミノヒドロラーゼ(N-acetylaminohydrolase)も使われる。この酵素は、尿素回路、フェニルアラニン代謝、トリプトファン代謝、シアノアミノ酸代謝などの代謝経路で活躍している。 3.1 遺伝子オントロジー(GO)コード 構造研究 As of late 2007, two structures have been solved for this class of enzymes, with PDB accession codes 2PLQ and 2UXY. 参考文献 IUBMB entry for 3.5.1.4(英語) BRENDA references for 3.5.1.4 (英語) PubMed references for 3.5.1.4(英語) PubMed Central references for 3.5.1.4(英語) Google Scholar references for 3.5.1.4(英語) Bray HG, James SP, Raffan IM, Ryman BE and Thorpe WV (1949). “The fate of certain organic acids and amides in the rabbit. 7. An amidase of rabbit liver”. Biochem. J. 44: 618–625. Bray HG, James SP, Thorpe WV and Wasdell MR (1950). “The fate of certain organic acids and amides in the rabbit. 11 Further observations on the hydrolysis of amides by tissue extracts”. Biochem. J. 47: 294–299. 外部リンク IUBMB entry for 3.5.1.4(英語) KEGG entry for 3.5.1.4(英語) BRENDA entry for 3.5.1.4(英語) NiceZyme view of 3.5.1.4(英語) EC2PDB: PDB structures for 3.5.1.4(英語) PRIAM entry for 3.5.1.4(英語) PUMA2 entry for 3.5.1.4(英語) IntEnz: Integrated Enzyme entry for 3.5.1.4(英語) MetaCyc entry for 3.5.1.4(英語) Atomic-resolution structures of enzymes belonging to this class(英語) 遺伝子オントロジー(GO)コード EGO entry for GO code 0004040: amidase AMIGO entry for GO code 0004040: amidase |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
アミノアシルtRNA合成酵素アミノアシルtRNA合成酵素 (aminoacyl-tRNA synthetase) とは、特定のアミノ酸 (またはその前駆体) その対応するtRNAにエステル結合させてアミノアシルtRNAを合成する酵素である。英語の略号としてaaRSやARSが用いられる。 アミノアシルtRNAは、リボソームに運ばれてtRNA部分の3塩基からなるアンチコドンが、mRNAのコーディング領域のコドンと対合し、タンパク質合成に用いられる。従って、3塩基のコドンと1アミノ酸の対応づけが行われる場はリボソームであっても、実際にコドンとアミノ酸の対応関係を示す遺伝暗号はaaRSの特異性にもとづいて規定されていることになる。 通常の生物では翻訳に使用されるアミノ酸20種類に対し、それぞれ対応するaaRSをもっている。例えば、アルギニンを認識してアルギニンtRNAにエステル結合する反応を触媒するaaRSはアルギニルtRNA合成酵素 (arginyl-tRNA synthetase) のように表記される。略号はArgRSのようにアミノ酸3文字表記+RSで表される。 起源RNAワールドにおいては、tRNA様のアダプター分子にRNA (リボザイム) がアミノ酸を結合させていたと考えられている。 現在の生物はほとんど共通する遺伝暗号を持っており、プロテインワールドの始原生物がリボザイムをタンパク質に置き換えつつ、アミノ酸とtRNAの対応関係は固定されたまま分子進化したと考えられる。そのため、aaRSは基質特異性を厳密に維持しつつも、最古のタンパク質として多様な進化を遂げている。その結果、3つの生物界 (真核生物、真正細菌、古細菌) の間で一次配列上の特徴が分かれていることが多い。 反応機構主反応aaRSは2段階の反応でATPの加水分解と共役してアミノ酸をtRNAに結合させる。 アミノ酸 + ATP → アミノアシルAMP + PPi (2) アミノアシルAMP-aaRS複合体に適切なtRNAが結合すると、アミノ酸のカルボキシ基がtRNAの3'末端のアデノシン (A76) の2'-または3'-OHとエステル結合を形成し、アデノシン一リン酸 (AMP) が遊離して反応が終結する。 アミノアシルAMP + tRNA → アミノアシルtRNA + AMP まとめると、アミノ酸 + ATP + tRNA → アミノアシルtRNA + AMP + PPi という反応式になる。 校正機構タンパク質合成に用いられるアミノ酸の中には、側鎖の大きさが似たアミノ酸が多く、単純にaaRS基質結合部位の形状を対応するアミノ酸をちょうど受け入れる形状にするだけでは十分な選択性が確保されない場合がある。この場合、本来特定のコドンに対応するアミノ酸ではないアミノ酸に翻訳されたタンパク質が一定割合で作られることになり、生物の生存に不都合である。 そこで、tRNAに本来とは異なるアミノ酸が結合 (ミスチャージ) した場合に、そのアミノアシルtRNAを加水分解する機構 (校正機構、editing) を有するaaRSが存在する。校正反応は多くの場合基質結合部位とは独立したドメイン (校正ドメイン) に加水分解に働く別の結合部位 (ミスチャージしたアミノ酸の側鎖と親和性が高い) で行われる。校正ドメインが別のポリペプチド鎖としてコードされている例も存在する。 分類aaRSは大きく2つのクラスに分けられる。それぞれのクラスはさらにIa、Ib、IcおよびIIa、IIb、IIcに分けられる。 クラスI
クラスII
アミノアシルtRNA合成酵素と遺伝暗号の拡張遺伝暗号の拡張の始まり始原生物が獲得した遺伝暗号に基づき、通常生物は20種類のアミノ酸 (標準アミノ酸) を翻訳で用いることができる。しかし、化学的に20種類以外のアミノ酸 (非天然型アミノ酸) をエステル結合したtRNAを試験管内翻訳系に加えると、リボソームは非天然型アミノ酸をタンパク質合成に使用し、tRNAのアンチコドンに対応するコドンに非天然型アミノ酸が対応付けられることが知られていた。このことから、既存のコドンに非天然型アミノ酸を対応付ける研究が行われるようになり、遺伝暗号の拡張 (Expansion of genetic code) と呼ばれるようになった。 遺伝暗号の拡張におけるaaRSの使用aaRSはアミノ酸とコドンの対応づけを行う酵素であるため、遺伝暗号の拡張のためにaaRSの基質特異性の改変を行うことが行われた。 一方、スクリプス研究所のピーター・シュルツらは2001年に同様の発想のもと、古細菌のチロシルtRNA合成酵素 (TyrRS) とアンバーサプレッサーtRNATyrを大腸菌内に導入し、大腸菌を用いた大規模スクリーニング法によって、古細菌TyrRSの基質特異性をO-メチルチロシンに高度に特異的なものにすることに成功した。これにより、その大腸菌の遺伝暗号はアンバーコドンにO-メチルチロシンが対応する遺伝暗号へと拡張したこととなる。 このスクリーニング法を用いることで、TyrRS変異体の中から種々の非天然型アミノ酸を特異的に認識するaaRSが選択され、一気にアミノ酸側から見た遺伝暗号の拡張が進んだ。 現在までの広がりさらに多くのaaRS-tRNAの組み合わせが同様の方法で遺伝暗号の拡張に用いることができることがわかっていった。特に近年では、ある古細菌で「22番目のアミノ酸」のピロリジンを認識し、アンバーコドンに対応するtRNAに結合させるピロリジルtRNA合成酵素 (PylRS) の基質認識が厳密でなく拡張容易なことを利用して、アセチルリジンやメチルリジンなど、生物の翻訳後修飾で生み出される側鎖を含む多くのアミノ酸の導入に成功している。その結果、遺伝暗号の拡張は従来の20種類のアミノ酸だけではできなかった細かなタンパク質の構造のチューニングや翻訳後修飾の遺伝子コード化など、実用性を増すこととなった。 関連項目リガーゼ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
アミラーゼアミラーゼ (amylase)とはジ(ヂ)アスターゼとも称される、膵液や唾液に含まれる消化酵素。グリコシド結合を加水分解することでデンプン(ラテン語"amylum")中のアミロースやアミロペクチンを、単糖類であるブドウ糖や二糖類であるマルトースおよびオリゴ糖に変換する酵素群である。 概要アミラーゼは1833年、フランスの生化学者、アンセルム・ペイアン (Anselme Payen) とジャン・ペルソー (Jean F. Persoz) が大麦の芽から取り出し、「切り離す」を意味するギリシア語の “διαστασις” より「ジ(ヂ)アスターゼ」と命名された。これが酵素の初めての単離である。 アミラーゼは消化酵素であり、デンプンやグリコーゲンを分解する。体内では主に、膵臓、耳下腺(唾液腺)から分泌され、またダイコンやカブ、ヤマイモにも多く含まれている。胃腸薬、消化剤として市販もされ、胃もたれや胸焼けの治療、防止に服用されている。 日本の製薬会社三共の事実上の創業者である高峰譲吉は、麹菌からジアスターゼを抽出し、自身の名の「タカ」を冠してタカジアスターゼと命名して1894年(明治27年)に特許を申請した[1]。高峰のジアスターゼ(アミラーゼ)の抽出成功は古くから餅を食べるとき大根おろしをつけて食べると胃がもたれないと言う事が大きなヒントとなったとも伝えられる。 夏目漱石の作品『吾輩は猫である』には、佐伯矩が発見した大根ジアスターゼについてと思われる新聞記事やタカジアスターゼを常用する人物が描写されて[2]、消化を促進するという機能が広く知られ用いられた様子がわかる。[3]。 現在、正式な物質名はアミラーゼであるが、旧名であるジアスターゼも医薬品/化学薬品の『タカジアスターゼ』として使用されている。 日本では現在も第一三共の医療用医薬品として「タカヂアスターゼ末」として薬局に卸されている(主に解熱鎮痛剤や整腸剤など他の散剤と混合して使うが、処方箋医薬品ではないため零売が可能)。また、第一三共ヘルスケアから一般用医薬品(胃腸薬)の「新タカヂア錠」と「第一三共胃腸薬」シリーズにタカヂアスターゼNとして配合されている。アメリカではパーク・デイビス(現:ファイザー)から市販されていない。 異性体α-アミラーゼ[4]、β-アミラーゼ[5]、グルコアミラーゼ[6]やイソアミラーゼ[7]がある。 α-アミラーゼ
α-アミラーゼは別名を1,4-α-D-グルカングルカノヒドロラーゼ、グリコゲナーゼといい、デンプンやグリコーゲンのα-1,4-結合を不規則に切断し、多糖ないしマルトース、オリゴ糖を生み出す酵素である。 β-アミラーゼ
β-アミラーゼは別名を1,4-α-D-グルカングルカノマルトヒドロラーゼ、グリコゲナーゼあるいはサッカロゲンアミラーゼといい、デンプンやグリコーゲンをマルトース(麦芽糖)に分解する。植物や微生物ではよく見られるが、動物からは見つかっていない。糖鎖の非還元末端から二つ目のα-1,4-グリコシド結合をエキソ型で逐次分解してマルトースを産生する。直鎖型のアミロースに対する分解効率は高い。一方、アミロペクチンに対してはα-1,6-グリコシド結合をしている分枝部で反応が停止し、マルトースとともにβリミットデキストリンが生成される。 グルコアミラーゼ
グルコアミラーゼは正式名称がグルカン1,4-α-グルコシダーゼといい、1,4-α-D-グルカングルコヒドロラーゼ、エキソ1,4-α-グルコシダーゼ、γ-アミラーゼ、リソソーマルα-グルコシダーゼあるいはアミログルコシダーゼを別名とする。糖鎖の非還元末端のα-1,4-結合をエキソ型に加水分解してブドウ糖1分子を産生する。α-1,6-結合も切断するものも知られている。 イソアミラーゼ詳細は「イソアミラーゼ」を参照 イソアミラーゼはアミロペクチンやグリコーゲン中のα-1,6-グリコシド結合を切断して直鎖のデキストリンやアミロースを生産する。ただし、プルランを分解できない。分枝部を切断するため、枝切り酵素や脱分枝酵素とも呼ばれる。 利用アミラーゼは、植物では果実の成熟や穀物の発芽の間に合成される。穀物酒や酢、水あめなどの伝統的な製法ではデンプンの糖化に麦芽に含まれるアミラーゼが用いられる。 微生物の分泌するアミラーゼは工業的に大量に生産され、製糖、食品加工、胃腸薬、衣料製造、洗剤等に利用されている。工業的にアミラーゼを生産する微生物としてはアスペルギルス・オリゼーや枯草菌が知られている。 尿中や血中のアミラーゼは、膵臓疾患や唾液腺疾患の診断に使われる。 ヒトアミラーゼヒトのアミラーゼには以下のものがある。
マクロアミラーゼ血症医療においてアミラーゼ高値を呈していることは、必ずしも膵疾患(特に急性・慢性膵炎)、唾液腺疾患を意味しない。疾患を合併しない代表的なものとしてマクロアミラーゼ血症がある。これはアミラーゼと免疫グロブリンが複合体を形成し、血清アミラーゼを測定すると高値を呈するもので、臓器障害を意味しない。 脚注
関連項目 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
DNAグリコシラーゼウラシルDNAグリコシラーゼの構造。 DNAグリコシラーゼ(DNA glycosylase、EC 3.2.2.3)は、DNAのN-グリコシド結合を加水分解する酵素の総称で、塩基除去修復において傷害のある塩基をDNAから取り除く役割を担う。チミンDNAグルコシラーゼ英語版、ウラシルDNAグルコシラーゼ英語版、オキソグアニングルコシラーゼ英語版など、傷害塩基の種類によってさまざまなものが存在する。反応の結果生じた塩基の無い部位(AP site)は、APエンドヌクレアーゼ、DNAリガーゼ等の塩基除去修復経路の下流の酵素によって処理される。 ウラシルDNAグルコシラーゼは、PCR産物間のコンタミネーションの防止のためにも使われている[1]。この他、ヒトの解糖系での反応を触媒する酵素の1つで、4量体で活性を示すグリセルアルデヒド-3-リン酸デヒドロゲナーゼの単量体が、ヒトのウラシルDNAグルコシラーゼと同じ物であることが判明した[2]。 出典2. ^ 『A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceroaldehyde-3-phospholate dehydrogenase.』 参考文献· IUBMB entry for 3.2.2.3(英語) · BRENDA references for 3.2.2.3 (英語) · PubMed references for 3.2.2.3(英語) · PubMed Central references for 3.2.2.3(英語) · Google Scholar references for 3.2.2.3(英語) 関連項目· 加水分解酵素 外部リンク]· IUBMB entry for 3.2.2.3(英語) · KEGG entry for 3.2.2.3(英語) · BRENDA entry for 3.2.2.3(英語) · NiceZyme view of 3.2.2.3(英語) · EC2PDB: PDB structures for 3.2.2.3(英語) · PRIAM entry for 3.2.2.3(英語) · PUMA2 entry for 3.2.2.3(英語) · IntEnz: Integrated Enzyme entry for 3.2.2.3(英語) · MetaCyc entry for 3.2.2.3(英語) · Atomic-resolution structures of enzymes belonging to this class(英語) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
アロステリック効果アロステリック効果(アロステリックこうか)とは、タンパク質の機能が他の化合物(制御物質、エフェクター)によって調節されることを言う。主に酵素反応に関して用いられる用語であるが、近年、Gタンパク質共役受容体 (GPCR) を中心とする受容体タンパク質の活性化制御において、アロステリック効果を示す化学物質 (アロステリックモジュレーター) の存在が知られるようになってきた。 アロステリー(allostery、その形容詞がアロステリックallosteric)という言葉は、ギリシア語で「別の」を意味するallosと「形」を意味するstereosから来ている。これは、一般にアロステリックタンパク質のエフェクターが基質と大きく異なる構造をしていることによる。このことから、制御中心が活性中心から離れた場所にあると考えられたのである。 しかし下記のヘモグロビンにおける酸素分子のように、同じ分子がエフェクターかつ基質となる例もあり、アロステリック効果は一般にヘモグロビンのようなオリゴマー構造でモデル化することができる(「アロステリック制御のモデル」の項参照)。 このため、アロステリック効果は タンパク質と化合物が一対多の複合体を形成する際に、前の段階の複合体形成によって次以降の複合体形成反応が促進・抑制されること、あるいはその複合体による反応が加速・減速されること。 と拡張定義されることも多い。 アロステリック制御アロステリック効果により主に酵素や受容体などのタンパク質の機能が制御される現象をアロステリック制御と呼ぶ。 酵素の場合、酵素の活性中心以外の部分(アロステリック部位)に対してエフェクター分子(反応に関係する物質でもそうでなくてもよい。)が会合して酵素のコンフォメーションが変化し、酵素の触媒活性や複合体形成反応の平衡定数が増減することを表す。 酵素の活性を促進するエフェクターはアロステリック・アクティベーターと呼ばれ、逆にタンパク質の活性を抑制するエフェクターはアロステリック・インヒビターと呼ばれる。アロステリック制御はフィードバック調節の一つの例である。 受容体の場合、内因性アゴニストのアゴニスト活性を促進するアロステリック部位に結合するリガンドはポジティブアロステリックモジュレーター (Positive Allosteric Modulator, PAM) と呼ばれ、逆にアゴニスト活性を抑制するアロステリックリガンドはネガティブアロステリックモジュレーター (Negative Allosteric Modulator, NAM) と呼ばれる。アロステリック部位に結合するだけで内因性アゴニストの活性に影響を与えないリガンドはサイレントアロステリックモジュレーター (Silent Allosteric Modulator, SAM) 、もしくはニュートラルアロステリックリガンド (Neutral Allosteric Ligand, NAL) と呼ばれる。 例血液中のヘモグロビンは酸素と結合する鉄中心を持つヘムを四つ持ち、各々の酸素との結合には一定の平衡定数が存在する。しかし、ヘモグロビン中の一つのヘムが酸素と結合を作るとヘモグロビン全体の構造が変化し、他のヘムと酸素との結合が促進される。すなわち、酸素濃度の高い所では単独のヘムよりも効率的に酸素を取り入れることができる。一方で、細胞中のミオグロビンのそれぞれのヘムにはヘモグロビンのような協同効果は無いので、酸素との結合生成反応は酸素濃度に一次で比例するだけである。この結果、ヘモグロビンは酸素の多い肺では酸素を吸収し、酸素の少ない各細胞では酸素を放出することができるのである。 アロステリック制御のモデル多くのアロステリック効果はジャック・モノー、ワイマン、ジャン・ピエール・シャンジューの唱える協奏モデルと、モノー・ワイマン・シャンジューモデルとダニエル・コシュランド、ネメシー、フィルマーの提唱する逐次モデルの両方で説明できる。どちらの説でも酵素サブユニットは緊張(T状態)か弛緩(R状態)のどちらかの状態にあると仮定し、弛緩状態のサブユニットは緊張状態のサブユニットよりも基質に結合しやすいとしている。二つのモデルは、サブユニット同士の関係と、両方の状態に至る前の状態に関する仮定の面で異なっている。 協奏モデルアロステリックに関する協奏モデルは対称モデルともモノー・ワイマン・シャンジュー (MWC) モデルとも呼ばれるが、一つのサブユニットの構造変化が他のサブユニットに影響を与えると仮定している。つまり、全てのサブユニットが同じコンフォメーションを取る。このモデルはリガンドがなくても成り立ち、T状態とR状態のコンフォメーションが均衡を保っている。一個のリガンド(もしくはアロステリックエフェクター)がアロステリック部位に結合すると、均衡はR状態もしくはT状態に移行する。 逐次モデルアロステリック制御の逐次モデルでは、一つのサブユニットのコンフォメーション変化が他のサブユニットに同様の変化を引き起こすとは考えない。つまり全てのサブユニットが同じコンフォメーションをとっている必要はない。さらに逐次モデルでは、基質分子が誘導適合モデルによって結合するとしている。一般的には、サブユニットがランダムに基質分子と衝突した時、活性中心が基質を包み込まなければならない。この誘導適合はサブユニットをT状態からR状態に移行させるが、近接サブユニットの構造を変化させることはない。その代わり、一つのサブユニットに基質が結合すると他のサブユニットの結合部位も基質に結合しやすいように徐々に構造を変えていく。要約すると、
アロステリック促進酸素分子がヘモグロビンに結合する時のように、アロステリック促進はリガンドの結合が基質分子と他の結合サイトの反応性を高める現象である。ヘモグロビンの例では、酸素は基質であると同時にエフェクターとして、効率的に働いている。アロステリックサイトは、隣のサブユニットの結合部位である。一つのサブユニットに酸素が結合すると、構造が変化し、残りの結合部位の酸素親和性を高める。 アロステリック抑制アロステリック抑制は、リガンドの結合によって結合部位の基質親和性が低下する現象である。例としては、2,3-ビスホスホグリセリン酸がヘモグロビンのアロステリック部位に結合すると、他の全てのサブユニットの酸素への親和性が低下する。 代謝系の生産物が、その系の中間反応を触媒する酵素の活性を抑制する場合、負のフィードバック制御の生体内における例であるとみなせるため、フィードバック阻害と呼ばれる。 エフェクターのタイプ多くのアロステリックタンパク質は自身の基質によって調節される。これらはホモトロピックアロステリック分子と呼ばれ、多くはアロステリック促進を示す。非基質の制御分子はヘテロトロピックアロステリック分子と呼ばれ、促進作用を示すものも抑制作用を示すものもある。自身の基質と非基質分子の両方で調節されるアロステリックタンパク質もある。このようなタンパク質はホモトロピック作用もヘテロトロピック作用も受ける。 関連項目
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
い イーディー=ホフステー図=ホフステー図(イーディー=ホフステーず、英: Eadie–Hofstee diagram)は、反応速度を反応速度と基質濃度の比の関数としてプロットする酵素反応速度論のグラフ表現である。ウルフ=イーディー=アウグスティンソン=ホフステープロット(Woolf–Eadie–Augustinsson–Hofstee plot)あるいはイーディー=アウグスティンソンプロットとも呼ばれる。
上式において、 これはミカエリス・メンテン式から以下のように導くことができる。 逆数を取り、 変形すると、
vをv/[S] に対してプロットすると、y切片としてVmax、x切片としてVmax/Km、負の傾きとしてKmが得られる。 ミカエリス・メンテン式を線型化するその他の手法と同様に、ヘインズ=ウルフプロットはKmやVmaxのような重要な反応速度論的パラメータを迅速に決定するために歴史的に使用されていたが、はるかに正確である非線型回帰手法に取って代わられている。ヘインズ=ウルフプロットはラインウィーバー=バークプロットよりも間違いが発生しやすいデータに対してより頑健である。これは、ヘインズ=ウルフプロットではいかなる範囲の基質濃度あるいは反応速度におけるデータ点も同等に重視するためである(ラインウィーバー=バークプロットはこのような点に対して不均等に重み付けをする)。どちらのプロットもデータをグラフを使って示す方法としては引き続き有用である。 ヘインズ=ウルフプロットの1つの欠点は、縦座標と横座標のどちらも独立変数を表わさない点である(どちらも反応速度に依存している)。そのため、全ての実験誤差がどちらの軸にも表われる。また、実験誤差あるいは不確かさが不均等に伝播し横座標の至るところでより大きくなり、それによってより小さなv/[S] の値により重視することになる。ゆえに、線型回帰の適合度の典型的な指標である相関係数Rを適用できない。 参考文献
関連項目 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EC番号EC番号(酵素番号、Enzyme Commission numbers)は酵素を整理すべく反応形式に従ってECに続く4組の数字で表したもの。 国際生化学連合(現在の国際生化学分子生物学連合)の酵素委員会によって1961年に作られた。 分類と命名法EC番号は酵素の系統的分類と関係が深い。また分類基準に共通項が存在するため、系統的命名法とEC番号とは少なからず対応関係を見出すことができる。 EC番号の分類基準は酵素の特性である反応特異性と基質特異性の違いにより区分されている。言い換えると、酵素反応の種類(反応特異性の違いを意味する)と基質の種類(基質特異性の違いを意味する)とで分類した番号である。 最初の数字が1であれば酸化還元酵素(オキシドレダクターゼ)で、2であれば転移酵素(トランスフェラーゼ)、3であれば、加水分解酵素(ヒドロラーゼ)、4であれば除去付加酵素(リアーゼ)、5であれば異性化酵素(イソメラーゼ)、6であれば合成酵素(リガーゼ、エピメラーゼ、ムターゼ、ラセマーゼ)となる。 さらに細かい反応特異性の違いや基質の違いにより番号が割り振られてゆく。分類は階層的でありECの接頭辞にピリオドで区切った続けた4個の番号 "EC X.X.X.X"(Xは数字)による表記がなされる。反応物質が二つ以上あるときはコロンで結ぶ場合もある。
全ての酵素についてこの番号が割り振られており、現在約 3,000 種類ほどの反応が見つかっている。またある活性を担う酵素が他の活性を有することも多く、ATPアーゼなどはATP加水分解反応のほかにタンパク質の加水分解反応への活性も持っている。 またEC番号は酵素を特定するのではなく、同じ基質に同じ反応で作用する酵素グループに対してEC番号が割り当てられることになる。つまりアイソザイムは同じEC番号を持つ。 命名法酵素の名前は国際生化学連合の酵素委員会によって命名される際に、同時にEC番号が与えられる。酵素の名称には「常用名」と「系統名」が付される。常用名と系統名の違いについて例をあげながら説明する: (例)次の酵素は全く同じ酵素(EC番号=EC 1.1.1.1)
系統名は、基質分子の名称(複数の場合は併記)と反応の名称を連結して命名される。系統名における反応の名称には規制があり、原則とし下記のいずれかが使用される: 常用名も、基本的には系統名と同じ規則で命名されるが、基質の一部を省略して短縮されたりしている。また、命名規則に従わない酵素も多く、DNAポリメラーゼなどはそのひとつである。 古くに発見され命名された酵素については、上述の規則ではなく当時の名称がそのまま使用されている。 などがこれにあたる。 以下に、EC番号と区分の対応を示す。 EC 1.-(酸化還元酵素)記事酸化還元酵素に詳しい
外部リンク
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
EC5050%効果濃度 (EC50)または半数効果濃度とは、薬物や抗体などが最低値からの最大反応の50%を示す濃度のことを指す[1]。 EC50は元来英語の"half maximal (50%) effective concentration"の略語であるが、現在ではEC50の表記の方が一般的である。 EC50は特に血中における50%効果濃度を指すこともある。この場合、投与量としてはED50(50%効果用量、半数効果用量)を用いる。 EC50は一般に医薬品の有効度を示すために用いられる。 効果の程度が連続的に計測できるような(例えば、心拍数に影響を及ぼすような)薬剤の用量反応曲線のEC50は、最大反応の50%の反応強度を示す化合物の濃度を表している [2]。 一方、効果が現れるか現れないかでのみ評価できる薬剤(例えば抗てんかん薬)の用量反応曲線のEC50は、試験された母集団のうち50%の個体が反応を示す化合物の濃度を表している [3]。 EC50は薬物の阻害能の尺度であるIC50とも関連している。競合結合評価と機能的アンタゴニスト評価のためにはIC50が用量反応曲線の最も一般的な集約尺度であり、アゴニスト/促進剤評価にはEC50が最も一般的である [4]。 用量反応曲線は通常、少しの濃度の変化に対して効果が急に立ち上がりやがて効果の増加が遅くなってゆく、いわゆるシグモイド曲線の形状を成す。濃度の増加に対して効果の増加が遅くなる点(変曲点)がIC50である。測定値に対して最適な近似曲線を数学的に導くことでIC50が求められる。 曲線回帰EC50を求めるための回帰曲線は薬剤の濃度(X)と効果の度合い(Y)の関数として以下のように表される。 Y = A + B − A 1 + ( X E C 50
) C {\displaystyle Y=A+{\frac {B-A}{1+({\frac {X}{EC_{50}}})^{C}}}} ここでAは測定値の最小値、Bは測定値の最大値、ヒル係数Cは勾配の最大値の絶対値を表す。 関連項目· 用量反応関係 · IC50 · 治療指数 脚注1. ^ Introducing doseresponse curves, Graphpad Software 2. ^ EC50 definition 3. ^ definition of EC50 for quantal dose response curve 4. ^ Assay Operations for SAR SupportNIH Chemical Genomics Center 外部リンク |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
イソプレン合成酵素
酵素の基質はジメチルアリル二リン酸であり、2つの生成物はイソプレンおよび二リン酸である。 この酵素はリアーゼファミリー、具体的にはリン酸エステルに作用する炭素-酸素リアーゼに属する。本酵素群の系統名はdimethylallyl-diphosphate diphosphate-lyase (isoprene-forming)である。一般的にはISPC、ISPSの略称が使われる。 推薦文献 Silver GM, Fall R (1991). “Enzymatic Synthesis of Isoprene from Dimethylallyl Diphosphate in Aspen Leaf Extracts”. Plant. Physiol. 97 (4): 1588–1591. doi:10.1104/pp.97.4.1588. PMC 1081206. PMID 16668590. Silver GM, Fall R (1995). “Characterization of aspen isoprene synthase, an enzyme responsible for leaf isoprene emission to the atmosphere”. J. Biol. Chem. 270 (22): 13010–6. doi:10.1074/jbc.270.22.13010. PMID 7768893. Wildermuth MC, Fall R (1996). “Light-Dependent Isoprene Emission (Characterization of a Thylakoid-Bound Isoprene Synthase in Salix discolor Chloroplasts)”. Plant. Physiol. 112 (1): 171–182. PMC 157936. PMID 12226383. Schnitzler JP, Arenz R, Steinbrecher R and Lehming A (1996). “Characterization of an isoprene synthase from leaves of Quercus petraea”. Bot. Acta 109: 216–221. Miller B, Oschinski C, Zimmer W (2001). “First isolation of an isoprene synthase gene from poplar and successful expression of the gene in Escherichia coli”. Planta. 213 (3): 483–7. doi:10.1007/s004250100557. PMID 11506373. Sivy TL, Shirk MC, Fall R (2002). “Isoprene synthase activity parallels fluctuations of isoprene release during growth of Bacillus subtilis”. Biochem. Biophys. Res. Commun. 294 (1): 71–5. doi:10.1016/S0006-291X(02)00435-7. PMID 12054742. Sasaki K, Ohara K, Yazaki K (2005). “Gene expression and characterization of isoprene synthase from Populus alba”. FEBS. Lett. 579 (11): 2514–8. doi:10.1016/j.febslet.2005.03.066. PMID 15848197. Schnitzler JP, Zimmer I, Bachl A, Arend M, Fromm J, Fischbach RJ (2005). “Biochemical properties of isoprene synthase in poplar (Populus x canescens)”. Planta. 222 (5): 777–86. doi:10.1007/s00425-005-0022-1. PMID 16052321. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
逸脱酵素 逸脱酵素(いつだつこうそ)とは、本来細胞内で働いている酵素が何らかの理由で血液中に流出したものである。 流出する理由としては、細胞自体の破壊、もしくは細胞膜の透過性亢進などで、多くの場合は組織障害に由来している。 臨床上、逸脱酵素の血中濃度を測定することで臓器がダメージを受けていないかを推測することが可能で、臨床検査の一環として頻繁に行われている。 また、一部の酵素は単に組織障害の指標となるだけでなく、それ自体が全身に障害を与える危険性を持っている。アミラーゼやリパーゼなどの、膵臓から逸脱する消化酵素がその代表である。 主な逸脱酵素[編集] AST(GOT) - ALT(GPT) - LDH - ALP - γ-GTP CK(CPK) - アミラーゼ - リパーゼ |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
インテグラーゼ インテグラーゼ(英: integrase)とはレトロウイルス(HIVを含む)により産生される酵素であり、感染細胞のDNAにレトロウイルスの遺伝物質を取り込ませることを可能にする。インテグラーゼは二本鎖DNAに組み込まれたウイルス(プロウイルス)によっても同じ目的のために産生される。 インテグラーゼはプレインテグレーション複合体(PIC)の重要な要素の1つである。 外部リンク Integrases - MeSH、米国国立医学図書館、生命科学用語シソーラス (英語サイト) http://discover.nci.nih.gov/pommier/IntegraseBookFull.pdf |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
サッカラーゼサッカラーゼ(saccharase)とは、スクロースをフルクトースとグルコースとに加水分解する酵素の一つである。IUPAC-IUB系統名はβ-D-フルクトフラノシダーゼ(E.C. 3.2.1.26)で別名としてインベルターゼあるいはインバーターゼ[1](invertase)、インベルチン(invertin)とも呼ばれる。この酵素によって生じたフルクトースとグルコースの混合物は転化糖と呼ばれる。 サッカラーゼはスクロースのフルクトースを認識して加水分解するのに対して、スクラーゼはグルコース側から分解する。したがって両者はスクロースを分解するが異なる酵素として認識されている。そして、サッカラーゼはスクロース以外にもラフィノースやスタキオース、フルクトオリゴ糖など、末端にフルクトフラノシド残基を含むオリゴ糖を基質とすることができ、中にはフルクトフラノシド残基を他の糖に転移させる活性を有する酵素も存在する。 サッカラーゼは、微生物、植物に広く分布しており、産業には細菌や酵母由来のものが利用されている。 CAZyにおける分類では、糖質加水分解酵素ファミリー32、68、100の3つのファミリーにわたって分類されている。 出典1. ^ NAID 110006453590 参考文献· 長倉三郎 ほか(編)「サッカラーゼ」『岩波理化学辞典』第5版 CD-ROM版、岩波書店、1998年。 関連項目· スクラーゼ · 転化糖 外部リンク· サッカラーゼ RCSC PDB(英語) サッカラーゼサッカラーゼ(saccharase)とは、スクロースをフルクトースとグルコースとに加水分解する酵素の一つである。IUPAC-IUB系統名はβ-D-フルクトフラノシダーゼ(E.C. 3.2.1.26)で別名としてインベルターゼあるいはインバーターゼ[1](invertase)、インベルチン(invertin)とも呼ばれる。この酵素によって生じたフルクトースとグルコースの混合物は転化糖と呼ばれる。 サッカラーゼはスクロースのフルクトースを認識して加水分解するのに対して、スクラーゼはグルコース側から分解する。したがって両者はスクロースを分解するが異なる酵素として認識されている。そして、サッカラーゼはスクロース以外にもラフィノースやスタキオース、フルクトオリゴ糖など、末端にフルクトフラノシド残基を含むオリゴ糖を基質とすることができ、中にはフルクトフラノシド残基を他の糖に転移させる活性を有する酵素も存在する。 サッカラーゼは、微生物、植物に広く分布しており、産業には細菌や酵母由来のものが利用されている。 CAZyにおける分類では、糖質加水分解酵素ファミリー32、68、100の3つのファミリーにわたって分類されている。 出典1. ^ NAID 110006453590 · スクラーゼ · 転化糖 外部リンク]· サッカラーゼ RCSC PDB(英語) カテゴリ: · EC 3.2.1 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
う ウイルス・ノイラミニダーゼ
インフルエンザウイルスの構造。ノイラミニダーゼは NA でヘマグルチニンは HA で示されている ウイルス・ノイラミニダーゼ (英: viral neuraminidase) は、インフルエンザウイルスの表面に存在するノイラミニダーゼの一種であり、宿主細胞内で産生された複製ウイルスの、細胞からの遊離を可能にする。ノイラミニダーゼは一般にシアル酸類を糖タンパク質から切断する酵素であり、インフルエンザウイルスの自己複製プロセスにおいて必要とされる。 インフルエンザウイルスが感染する際、ウイルスの表面にあるヘマグルチニン (シアル酸類に結合する分子) を使って宿主細胞に吸着する。シアル酸類は、宿主細胞の糖タンパク質に広く見出される糖 (9炭糖)であり、ウイルスはこの分子グループを悪用して宿主細胞に結合する。宿主細胞からウイルスが遊離されるためには、ノイラミニダーゼが酵素として特異的に、ヘマグルチニンが結合する宿主の糖タンパク質のシアル酸を切断しなければならない[2]。インフルエンザの自己複製プロセスの不可欠なパートとして、ノイラミニダーゼの機能をノイラミニダーゼ阻害剤でブロックすることは、インフルエンザ治療の有効な方法である。 ムンプスウイルス(流行性耳下腺炎の病原体)およびヒトパラインフルエンザウイルスを含むいくつかのウイルス群においては、ヘマグルチニン-ノイラミニダーゼタンパク質1つで、ノイラミニダーゼとヘマグルチニンの両方の機能を果たしている。
機能
インフルエンザウイルスの自己複製。ステップ 6と7で、ノイラミニダーゼとヘマグルチニンと宿主細胞の細胞膜が結合してウイルスを包む膜であるエンベロープを構成し、これが個々の複製ウイルス本体を包み込みながら「出芽」して宿主細胞から遊離される様子を示している。 この酵素は、複製されたインフルエンザウイルスの宿主細胞からの遊離を促進する。インフルエンザウイルスのエンベロープはヘマグルチニンとノイラミニダーゼという2種類の糖タンパク質を持つ。ヘマグルチニンはウイルスが宿主に感染する場合に必要であるが、宿主内で複製されたウイルスが出芽により遊離される際には、複製されたウイルスが宿主細胞の表面にあるシアル酸に、それ自身のヘマグルチニンで結合したままとなり、遊離が阻害される。ウイルス・ノイラミニダーゼは、宿主細胞の表面にある糖タンパク質からシアル酸残基を切断して、ヘマグルチニンと宿主細胞の糖タンパク質との結合を切り離し、子孫ウイルスを遊離させ、宿主周辺の非感染細胞への感染を可能にする[3]。また、ノイラミニダーゼは、ウイルス表面にある糖タンパク質のシアル酸残基を切断することで、ウイルス同士が凝集することも防いでいる。 ノイラミニダーゼ阻害剤]ノイラミニダーゼは、「構造に基づく酵素阻害剤の設計プログラム」のターゲットにされて来た。このプログラムは、ザナミビル (リレンザ) および オセルタミビル (タミフル)という2種類の薬剤の開発という成果を上げている。ノイラミニダーゼ阻害剤の投与は、症状のひどさを緩和し、ウイルスの蔓延を抑える治療法である。ザナミビルの投与は吸入により、オセルタミビルの投与は経口により行なわれる。2010年1月に日本において製造販売承認を取得したペラミビル(ラピアクタ)は注射(点滴静注)[4]により投与される。世界に先駆けて日本で、2010年10月に純国産の吸入ノイラミニダーゼ阻害薬であるラニナミビル製剤「イナビル®吸入粉末剤20mg」が発売された。 ノイラミターゼ阻害剤耐性2005年2月27日に、14歳のベトナム人の少女が、インフルエンザの治療で使用されているオセルタミビルに対して耐性を持つ H5N1 インフルエンザウイルスの変異株に感染していたことが報告されている[5] 。彼女は予防的な投与 (75 mg 1日1回) を受けていたのであるが、この治療法に対する感受性が無かったのである。鳥類による媒介でインフルエンザの世界的流行の脅威が高まる中で、科学者達はタミフル治療への耐性の発生原因の探索を開始し、耐性ウイルスのノイラミニダーゼの274番目のアミノ酸が通常のヒスチジンからチロシンに置き変わっていたことが原因であったことを突き止めている。この変異はザナミビルとラニナミビルに対しては耐性を獲得しないことが判明している。 インフルエンザの変異株は突然変異により継続的に出現することから、薬剤耐性の原因である、ノイラミニダーゼのサブタイプの決定を、科学者達が迅速かつ効果的に行なうことが、特定の種類のインフルエンザ変異株と闘うためには不可欠である。 基質特異性理想的には、インフルエンザのウイルス・ノイラミニダーゼ (NA) は、ヘマグルチニン (HA) のレセプター(糖タンパク質)と同じタイプのレセプターにのみ作用すべきであるが、これが常に実現するわけではない。NA と HA の基質特異性に強い一致が無い場合に、ウイルスがいったいどうやって切り抜けているのかは全く不明である。 エキソ型(exo)とエンド型(endo)ノイラミニダーゼ酵素群はエキソ型(exo)またはエンド型(endo)のグリコシダーゼ 活性を持つことができる。これは EC 3.2.1.29 (endo-neuraminidase) [6] および EC 3.2.1.18 (exo-neuraminidases)[7]で分類されている。一般に哺乳類のシアル酸残基は、グリカン複合体の終端部に位置し、ウイルス・ノイラミニダーゼはエキソ型グリコシダーゼであり、これらの終端の残基を自分達の基質と認識して作用する(グリコシダーゼ#分類を参照)。 脚注1. ^ Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA, Colman PM (November 1992), “The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor”, Proteins 14 (3): 327–32, doi:10.1002/prot.340140302, PMID 1438172 2. ^ Huang IC, Li W, Sui J, Marasco W, Choe H, Farzan M (May 2008), “Influenza A virus neuraminidase limits viral superinfection”, J. Virol. 82 (10): 4834–43, doi:10.1128/JVI.00079-08, PMC 2346733, PMID 18321971 3. ^ 河岡義裕・堀本研子 『インフルエンザパンデミック』 講談社〈ブルーバックス〉、2009年、51頁。 4. ^ “抗インフルエンザウイルス剤「ラピアクタ点滴用バッグ300mg」および「ラピアクタ点滴用バイアル150mg」の製造販売承認取得について” (PDF) (プレスリリース), 塩野義製薬, (2010年1月13日) 2010年12月24日閲覧。 5. ^ USATODAY.com - Vietnamese girl's bird flu infection resistant to Tamiflu 6. ^ EC 3.2.1.129 7. ^ EC 3.2.1.18 外部リンク
関連項目 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
DNAグリコシラーゼウラシルDNAグリコシラーゼの構造。 DNAグリコシラーゼ(DNA glycosylase、EC 3.2.2.3)は、DNAのN-グリコシド結合を加水分解する酵素の総称で、塩基除去修復において傷害のある塩基をDNAから取り除く役割を担う。チミンDNAグルコシラーゼ英語版、ウラシルDNAグルコシラーゼ英語版、オキソグアニングルコシラーゼ英語版など、傷害塩基の種類によってさまざまなものが存在する。反応の結果生じた塩基の無い部位(AP site)は、APエンドヌクレアーゼ、DNAリガーゼ等の塩基除去修復経路の下流の酵素によって処理される。 ウラシルDNAグルコシラーゼは、PCR産物間のコンタミネーションの防止のためにも使われている[1]。この他、ヒトの解糖系での反応を触媒する酵素の1つで、4量体で活性を示すグリセルアルデヒド-3-リン酸デヒドロゲナーゼの単量体が、ヒトのウラシルDNAグルコシラーゼと同じ物であることが判明した[2]。 出典2. ^ 『A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceroaldehyde-3-phospholate dehydrogenase.』 参考文献· IUBMB entry for 3.2.2.3(英語) · BRENDA references for 3.2.2.3 (英語) · PubMed references for 3.2.2.3(英語) · PubMed Central references for 3.2.2.3(英語) · Google Scholar references for 3.2.2.3(英語) 関連項目· 加水分解酵素 外部リンク]· IUBMB entry for 3.2.2.3(英語) · KEGG entry for 3.2.2.3(英語) · BRENDA entry for 3.2.2.3(英語) · NiceZyme view of 3.2.2.3(英語) · EC2PDB: PDB structures for 3.2.2.3(英語) · PRIAM entry for 3.2.2.3(英語) · PUMA2 entry for 3.2.2.3(英語) · IntEnz: Integrated Enzyme entry for 3.2.2.3(英語) · MetaCyc entry for 3.2.2.3(英語) · Atomic-resolution structures of enzymes belonging to this class(英語) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
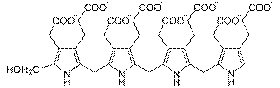
ウロポルフィリノーゲンIIIシンターゼ
ウロポルフィリノーゲンIIIシンターゼ(Uroporphyrinogen III synthase)(ウロポルフィリノーゲンIII合成酵素)とは、ヒドロキシメチルビランをウロポルフィリノーゲンIIIに変換するポルフィリン代謝の第4番目の反応に関わる酵素である。 ヒドロキシメチルビランがウロポルフィリノーゲンIIIシンターゼによって縮合し、環を巻くとウロポルフィリノーゲンIIIとなる。この際、ウロポルフィリノーゲンIIIシンターゼの働きにより4つのピロール環が整然と並んだヒドロキシメチルビランの一端のピロール環一つだけが反転して縮合し環を形成する。ウロポルフィリノーゲンIIIシンターゼがはたらかない場合、ピロール環が整然と並んだままのヒドロキシメチルビランが自発的に縮環してウロポルフィリノーゲンI が生成する。ウロポルフィリノーゲンI はウロポルフィリノーゲン脱炭酸酵素の基質となりコプロポルフィリノーゲンIへと変換されるが、これはコプロポルフィリノーゲン酸化酵素の基質とならないため、プロトポルフィリンには至らない。[1]。 ⇒
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||





|
ウロポルフィリノーゲンIII |
|
|
|
|
|
識別情報 |
|
|
特性 |
|
|
C40H44N4O16 |
|
|
836.795 g/mol |
|

ウロポルフィリノーゲンIII(Uroporphyrinogen III) は、ポルフィリンの生合成において、ヒドロキシメチルビランからウロポルフィリノーゲンIIIシンターゼ(合成酵素)により作られ、ウロポルフィリノーゲンIII脱カルボキシ酵素によりコプロポルフィリノーゲンIIIに変換される。

δ-アミノレブリン酸からプロトポルフィリンIXまでの生合成経路

ヘム合成は、細胞質やミトコンドリア内(黄色部分)で反応が起こる。
ヒドロキシメチルビラン ウロポルフィリノーゲンIII
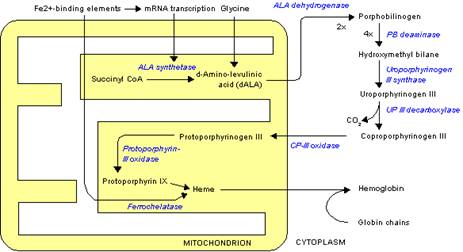
ヘム合成は、細胞質やミトコンドリア内(黄色部分)で反応が起こる。
病理学
この酵素の欠乏は、ガンサー病(en:Gunther's disease)を引き起こす。
脚注
^ はじめに: ポルフィリン症 メルクマニュアル18版 日本語版
関連項目
ポルフィリン症
プロトポルフィリン
え
セルラーゼ
セルラーゼ (Cellulase) とは、β-1,4-グルカン(例えば、セルロース)のグリコシド結合を加水分解する酵素。主に細菌や植物において作られ、生物界に広く存在する。
分子内部から切断するエンドグルカナーゼ EC 3.2.1.4 と、糖鎖の還元末端と非還元末端のいずれから分解し、セロビオースを遊離するエキソグルカナーゼ(セロビオヒドロラーゼ) EC 3.2.1.91 にわけられる。また酵素タンパク質の構造から、ファミリーに分けられている。
保有生物
菌類など生産能を有している生物のほか、哺乳類では体内に生産能を持つ別の生物を共生させているものがある。
動物類
貝類
動物では巻き貝や二枚貝がセルラーゼ、ヘミセルラーゼを産生できる。
節足動物門
シロアリやゴキブリはセルラーゼを産生する単細胞の原生生物を腸内に共生させている。動物自身はセルラーゼを産生できないためこのような共生をおこなっていると考えられてきたが、シロアリの研究では、シロアリ自身のゲノムにセルラーゼをコードする遺伝子が存在し、この遺伝子が共生するバクテリアや原生生物から近年に水平転移したものでは無いことが示唆されている (Watanabe et al. 1998)。マツノザイセンチュウもセルラーゼ遺伝子の発現が認められるという報告がある。
深海底に生息するカイコウオオソコエビでは、高いグルコース(ブドウ糖)生産性を有している。
哺乳類
ウシやヒツジなどの反芻動物やウマなどの草食動物は消化管にセルラーゼを産生する微生物(細菌、糸状菌、原生生物)を生息させており、これらによるセルロース分解によって植物繊維の消化を可能にしている。
菌類
子嚢菌類、担子菌類にはセルロース分解能を持つものが多い。木材の分解はこれらが主体となっており、木材腐朽菌と言われる。糸状菌トリコデルマの1種 Trichoderma reesei はセルラーゼ高生産菌として有名な菌である。50~60 g/lのタンパク質を分泌し、その大部分がセルラーゼ、ヘミセルラーゼを占めている。少なくとも5種のエンドグルカナーゼと2種類のセロビオハイドロラーゼといった複数のセルラーゼを生産することが分かっており、セルロース分解において期待されている。
好熱嫌気性セルロース分解細菌 Clostridium thermocellum では複数のサブユニットからなるセルラーゼ複合体 — セルロソーム (Cellulosome) を形成していることが知られており、これが高いセルロース分解能につながっていると考えられている。
応用
植物細胞の細胞壁のみを分解し、植物細胞のプロトプラスト化する場合や、繊維の間の汚れを取るために市販の洗剤に配合されたり、ジーンズ繊維の材質の改善などに使われている。また、カイコウオオソコエビ由来のセルラーゼは廃材などのセルロースを常温でグルコース(ブドウ糖)に変換できることから、穀物を原料としないアルコール燃料の生産に寄与することが期待されている[1]。
関連項目
· 加水分解酵素
参考文献
· H Watanabe et al. Nature 394, 330-331, 1998
1. ^ マリアナ海溝世界最深部に生息する超深海性ヨコエビの特異な生態の解明と新規セルラーゼの発見 海洋研究開発機構
カテゴリ:
ヌクレアーゼ
ヌクレアーゼ(Nuclease)は核酸分解酵素の総称。デオキシリボ核酸ないしリボ核酸の糖とリン酸の間のホスホジエステル結合を加水分解してヌクレオチドとする。
RNAを分解するリボヌクレアーゼとDNAを分解するデオキシリボヌクレアーゼに分類できる他、両方を分解することができるヌクレアーゼも知られており、その役割も様々である。ウイルスが有するヌクレアーゼには宿主の核酸を分解して自らの核酸の原料とする役割をもつものがある。また、制限酵素もヌクレアーゼの一種であり、これは外来の核酸を分解してウイルスの感染、増殖を防ぐ役割があると考えられている。核酸がメチル化されているとヌクレアーゼは働かなくなるため、自分の核酸を無闇に分解しないようにこの酵素を有する細菌も多い。多細胞生物においては死滅した細胞の核酸を分解するためにヌクレーゼが生産されることがあるほか、特殊な例としては紫外線などの影響で二量化したチミジンをとりはずすためのヌクレアーゼが存在する。
分解の型式により、エンドヌクレアーゼとエキソヌクレアーゼという分類もできる。
エンドヌクレアーゼ(英:endonuclease)
核酸配列の内部(endo-)で核酸を切断する酵素で、すなわち糸を途中で切るように核酸を切断する。制限酵素は代表的なエンドヌクレアーゼである。
エキソヌクレアーゼ(英:exonuclease)
核酸配列の外側(exo-)から、すなわち核酸の5'端または3'端から削るように分解する。DNAポリメラーゼにもエキソヌクレアーゼ活性があるが、それはDNA複製中のミスを校正するためであると考えられている。
枝切り酵素
枝切り酵素(debranching enzyme)は、枝切りアミロース、デブランチングエンザイムとも呼ばれる。澱粉やグリコーゲンのα-1,6グルコシド結合を加水分解し、直鎖状のアミロースを生成する酵素の総称[1]。この中にイソアミラーゼやプルラナーゼ、α-1,6グルコシダーゼ、R-酵素等が含まれる[2]。
産業小史
1973年、A.E.ステイラー社が澱粉液化液にグルコアミラーゼとα-1,6グルコシダーゼを用いて糖化を行うと、グルコースのブドウ糖収量がアップすることを発表。
1978年、天野製薬(現・天野エンザイム)がBacillus sectramas起源のプルラナーゼを開発。
1979年、通産省工業技術院発酵研究所(現・産業技術総合研究所)の高崎義幸が、Bacillus sectramas由来のα-1,6グルコシダーゼを澱粉糖化時にグルコアミラーゼと共存させると、ブドウ糖の収率が上がることを発表。
1981年、林原 (企業) がグルコアミラーゼによる澱粉糖化時にイソアミラーゼを併用し、ブドウ糖収率が上がることを発表。
1981年、ノボインダストリージャパンが性質を向上させたプルラナーゼを開発したと発表。Bacillus acidopulluliticus由来、商品名「ノボザイム」。
1992年、天野製薬が耐熱性・耐酸性の向上したプルラナーゼを開発。バチルス属由来、商品名「シルバーラーゼ」(グルコアミラーゼとの混合品)。
1992年、ナガセ生化学工業(現・ナガセケムテックス)がBacillus circulans由来のプルラナーゼを開発。
1995年、大和化成がBacillus brevis由来の枝切り酵素を開発。
ソルベイ社(ジェネンコア社、現・ダニスコ社)が、組換え菌によるプルラナーゼを開発。Bacillus deramificansの産生するプルラナーゼの遺伝子をBacillus licheniformisに導入。2001年に日本国内で発売開始[2]。
関連項目
|
プルラナーゼ(EC 3.2.1.41) |
イソアミラーゼ(EC 3.2.1.68) |
出典
^ 『澱粉の科学と技術』 ISBN 978-4990528706
^ a b 「日本酵素産業小史」日本酵素協会、2009年5月
エロスルファーゼ アルファ
|
臨床データ |
販売名 |
Vimizim |
Drugs.com |
Multum Consumer Information |
胎児危険度分類
|
US: C |
|
法的規制 |
US: ℞-only |
識別 |
|
CAS番号 |
9025-60-9 |
ATCコード |
A16AB12 (WHO) |
IUPHAR/BPS |
|
7392 |
ChemSpider |
none |
化学的データ |
化学式 |
|
C5020H7588N1364O1418S34 |
分子量 |
110.8 kg/mol |
|
|
エロスルファーゼ アルファ(Elosulfase alfa)はムコ多糖症IV-A型(モルキオ症候群)の治療に用いられる医薬品である。遺伝子組み換えによって合成されるN-アセチルガラクトサミン-6-スルファターゼ(GALNS)であり、モルキオ症候群への酵素補充療法に用いられる。商品名ビミジム。
米国で2014年2月に認可された[1]。日本では2014年12月に承認された[2]。
エロスルファーゼ アルファは酵素補充療法に用いられる医薬品であり、2014年の臨床研究で若年のモルキオ症候群A型患者に対して効果があることが示された[3]。この治療は呼吸器症状、日常生活動作、成長等に有効性を示す事が2015年の論文で示されている[4]。
副作用
治験では72.4%に副作用が見られ、主な副作用は、Infusion associated reaction(65.5%)、発熱(32.8%)、嘔吐(31.0%)、頭痛(25.9%)、悪心(24.1%)であった[5]。
重大な副作用は、Infusion associated reaction(アナフィラキシー)である。
出典
^ “FDA approves Vimizim to treat rare congenital enzyme disorder”. Food and Drug Administration (2014年2月14日). 2015年2月19日閲覧。
^ “新薬14製品が承認 8製品が難病等患者数の少ない疾患向け”. ミクス (2014年12月26日). 2015年2月19日閲覧。
^ Hendriksz, C. et. al (November 2014). “Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study”. Journal of Inherited Metabolic Disease 37 (6): 979-990. doi:10.1007/s10545-014-9715-6.
^ Hendriksz, C. et. al (February 2015). “Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial”. Molecular Genetics and Metabolism 114 (2): 178–185. doi:10.1016/j.ymgme.2014.08.012.
^ “ビミジム点滴静注液5mg 添付文書” (2015年11月). 2016年7月7日閲覧。
カテゴリ: ペプチド酵素EC 3.1.6希少疾病用医薬品
希少疾病用医薬品カテゴリ「希少疾病用医薬品」にあるページこのカテゴリには 37 ページが含まれており、そのうち以下の 37 ページを表示しています。
あ
い
え
おか
く
こ
さ
す
せ
ち
と
に
ぬ
は
へ
ほ
み
り
れ
ろ
|
セルラーゼ
セルラーゼ (Cellulase) とは、β-1,4-グルカン(例えば、セルロース)のグリコシド結合を加水分解する酵素。主に細菌や植物において作られ、生物界に広く存在する。
分子内部から切断するエンドグルカナーゼ EC 3.2.1.4 と、糖鎖の還元末端と非還元末端のいずれから分解し、セロビオースを遊離するエキソグルカナーゼ(セロビオヒドロラーゼ) EC 3.2.1.91 にわけられる。また酵素タンパク質の構造から、ファミリーに分けられている。
保有生物
菌類など生産能を有している生物のほか、哺乳類では体内に生産能を持つ別の生物を共生させているものがある。
動物類
貝類
動物では巻き貝や二枚貝がセルラーゼ、ヘミセルラーゼを産生できる。
節足動物門
シロアリやゴキブリはセルラーゼを産生する単細胞の原生生物を腸内に共生させている。動物自身はセルラーゼを産生できないためこのような共生をおこなっていると考えられてきたが、シロアリの研究では、シロアリ自身のゲノムにセルラーゼをコードする遺伝子が存在し、この遺伝子が共生するバクテリアや原生生物から近年に水平転移したものでは無いことが示唆されている (Watanabe et al. 1998)。マツノザイセンチュウもセルラーゼ遺伝子の発現が認められるという報告がある。
深海底に生息するカイコウオオソコエビでは、高いグルコース(ブドウ糖)生産性を有している。
哺乳類
ウシやヒツジなどの反芻動物やウマなどの草食動物は消化管にセルラーゼを産生する微生物(細菌、糸状菌、原生生物)を生息させており、これらによるセルロース分解によって植物繊維の消化を可能にしている。
菌類
子嚢菌類、担子菌類にはセルロース分解能を持つものが多い。木材の分解はこれらが主体となっており、木材腐朽菌と言われる。糸状菌トリコデルマの1種 Trichoderma reesei はセルラーゼ高生産菌として有名な菌である。50~60 g/lのタンパク質を分泌し、その大部分がセルラーゼ、ヘミセルラーゼを占めている。少なくとも5種のエンドグルカナーゼと2種類のセロビオハイドロラーゼといった複数のセルラーゼを生産することが分かっており、セルロース分解において期待されている。
好熱嫌気性セルロース分解細菌 Clostridium thermocellum では複数のサブユニットからなるセルラーゼ複合体 — セルロソーム (Cellulosome) を形成していることが知られており、これが高いセルロース分解能につながっていると考えられている。
応用
植物細胞の細胞壁のみを分解し、植物細胞のプロトプラスト化する場合や、繊維の間の汚れを取るために市販の洗剤に配合されたり、ジーンズ繊維の材質の改善などに使われている。また、カイコウオオソコエビ由来のセルラーゼは廃材などのセルロースを常温でグルコース(ブドウ糖)に変換できることから、穀物を原料としないアルコール燃料の生産に寄与することが期待されている[1]。
関連項目
· 加水分解酵素
参考文献
· H Watanabe et al. Nature 394, 330-331, 1998
1. ^ マリアナ海溝世界最深部に生息する超深海性ヨコエビの特異な生態の解明と新規セルラーゼの発見 海洋研究開発機構
ヌクレアーゼ
ヌクレアーゼ(Nuclease)は核酸分解酵素の総称。デオキシリボ核酸ないしリボ核酸の糖とリン酸の間のホスホジエステル結合を加水分解してヌクレオチドとする。
RNAを分解するリボヌクレアーゼとDNAを分解するデオキシリボヌクレアーゼに分類できる他、両方を分解することができるヌクレアーゼも知られており、その役割も様々である。ウイルスが有するヌクレアーゼには宿主の核酸を分解して自らの核酸の原料とする役割をもつものがある。また、制限酵素もヌクレアーゼの一種であり、これは外来の核酸を分解してウイルスの感染、増殖を防ぐ役割があると考えられている。核酸がメチル化されているとヌクレアーゼは働かなくなるため、自分の核酸を無闇に分解しないようにこの酵素を有する細菌も多い。多細胞生物においては死滅した細胞の核酸を分解するためにヌクレーゼが生産されることがあるほか、特殊な例としては紫外線などの影響で二量化したチミジンをとりはずすためのヌクレアーゼが存在する。
分解の型式により、エンドヌクレアーゼとエキソヌクレアーゼという分類もできる。
エンドヌクレアーゼ(英:endonuclease)
核酸配列の内部(endo-)で核酸を切断する酵素で、すなわち糸を途中で切るように核酸を切断する。制限酵素は代表的なエンドヌクレアーゼである。
エキソヌクレアーゼ(英:exonuclease)
核酸配列の外側(exo-)から、すなわち核酸の5'端または3'端から削るように分解する。DNAポリメラーゼにもエキソヌクレアーゼ活性があるが、それはDNA複製中のミスを校正するためであると考えられている。
お
β-ラクタマーゼ
β-ラクタマーゼ(ベータラクタマーゼ、β-lactamase)とはβ‐ラクタム系抗生物質を加水分解する酵素である。ペニシリン/セファロスポリンアミド-β-ラクタムヒドロラーゼ (penicillin/cepharosporin amido-β-lactam hydrolase)とも呼ばれる。EC3.5.2.6に分類される酵素である。
幾つかの種類のグラム陰性菌がβ-ラクタマーゼを産生することでβ-ラクタムに対して耐性を示すことが知られている。なお、β-ラクタム耐性はβ-ラクタマーゼのみが原因ではなくMRSAのようにペニシリン結合タンパク質の基質特異性が変化しても現れる。
現在β-ラクタマーゼは基質特異性の違いにより
· ペニシリナーゼ (クラスA β-ラクタマーゼ)
· メタロ-β-ラクタマーゼ (クラスB β-ラクタマーゼ、亜鉛-β-ラクタマーゼ、カルバペネマーゼ)
· セファロスポリナーゼ (クラスC β-ラクタマーゼ)
· オキサシリナーゼ (クラスD β-ラクタマーゼ)
これら4種のβ-ラクタマーゼのうち、クラスB β-ラクタマーゼは活性中心に亜鉛を持つが、他はセリン残基を持つ。ペニシリナーゼはペニシリン系抗生物質と第二世代セファロスポリンを分解するのに対して、セファロスポリナーゼは主にセファロスポリンを分解する。オキサシリナーゼはオキサシリンをも分解するペニシリナーゼであり、メタロ-β-ラクタマーゼはカルバペネム系抗生物質を分解する点に特徴がある。
β-ラクタマーゼの遺伝子は、細菌の染色体上あるいはプラスミド上に存在する。特に伝達性薬剤耐性プラスミド (drug resistance plasmid)に存在するβ-ラクタマーゼ遺伝子は菌種特異性も少なく多剤耐性菌の発生にも関与していると考えられる。
脚注
1. ^ β-ラクタム耐性菌とその検出方法、関東化学
2. ^ Bush, K. et. al. A functional classification scheme for β-lactamases and its correlation with molecular structure, Antimicrob Agents Chemother., 39, 1211-1233, 1995.
3. ^ Ambler, R. P., The structure of β-lactameses, Philos Trans R Society Lond (Biol), 289, 321-331, 1980.
4. ^ 石井良和、基質特異性拡張型β-ラクタマーゼ産生大腸菌、クレブシエラ、臨床と微生物、26、121-125, 1999.
関連項目
· クラブラン酸
· 薬剤耐性
· ペニシリン
· セファロスポリン
出典
· β-ラクタマーゼ『生物学辞典』第4版、岩波書店。
· β-ラクタマーゼについて 日本ベクトン・ディッキンソン株式会社
β-ラクタマーゼ
β-ラクタマーゼ(ベータラクタマーゼ、β-lactamase)とはβ‐ラクタム系抗生物質を加水分解する酵素である。ペニシリン/セファロスポリンアミド-β-ラクタムヒドロラーゼ (penicillin/cepharosporin amido-β-lactam hydrolase)とも呼ばれる。EC3.5.2.6に分類される酵素である。
幾つかの種類のグラム陰性菌がβ-ラクタマーゼを産生することでβ-ラクタムに対して耐性を示すことが知られている。なお、β-ラクタム耐性はβ-ラクタマーゼのみが原因ではなくMRSAのようにペニシリン結合タンパク質の基質特異性が変化しても現れる。
現在β-ラクタマーゼは基質特異性の違いにより
· ペニシリナーゼ (クラスA β-ラクタマーゼ)
· メタロ-β-ラクタマーゼ (クラスB β-ラクタマーゼ、亜鉛-β-ラクタマーゼ、カルバペネマーゼ)
· セファロスポリナーゼ (クラスC β-ラクタマーゼ)
· オキサシリナーゼ (クラスD β-ラクタマーゼ)
これら4種のβ-ラクタマーゼのうち、クラスB β-ラクタマーゼは活性中心に亜鉛を持つが、他はセリン残基を持つ。ペニシリナーゼはペニシリン系抗生物質と第二世代セファロスポリンを分解するのに対して、セファロスポリナーゼは主にセファロスポリンを分解する。オキサシリナーゼはオキサシリンをも分解するペニシリナーゼであり、メタロ-β-ラクタマーゼはカルバペネム系抗生物質を分解する点に特徴がある。
β-ラクタマーゼの遺伝子は、細菌の染色体上あるいはプラスミド上に存在する。特に伝達性薬剤耐性プラスミド (drug resistance plasmid)に存在するβ-ラクタマーゼ遺伝子は菌種特異性も少なく多剤耐性菌の発生にも関与していると考えられる。
脚注
1. ^ β-ラクタム耐性菌とその検出方法、関東化学
2. ^ Bush, K. et. al. A functional classification scheme for β-lactamases and its correlation with molecular structure, Antimicrob Agents Chemother., 39, 1211-1233, 1995.
3. ^ Ambler, R. P., The structure of β-lactameses, Philos Trans R Society Lond (Biol), 289, 321-331, 1980.
4. ^ 石井良和、基質特異性拡張型β-ラクタマーゼ産生大腸菌、クレブシエラ、臨床と微生物、26、121-125, 1999.
関連項目
· クラブラン酸
· 薬剤耐性
· ペニシリン
· セファロスポリン
出典
· β-ラクタマーゼ『生物学辞典』第4版、岩波書店。
· β-ラクタマーゼについて 日本ベクトン・ディッキンソン株式会社
酸素添加酵素
酸素添加酵素(さんそてんかこうそ、oxygenase)とは、酸化還元酵素の一種で、分子状酸素の基質への直接の取り込み反応を触媒する。酸素原子を挿入する酸化反応を触媒する酵素である。オキシゲナーゼ、酸素化酵素とも呼ばれる。
酸素添加酵素は酸素分子を利用するが、酸素分子の二つの酸素原子を基質と結合させる二酸素添加酵素(dioxygenase)と、一方の酸素を基質と結合させるが他方は、水素を添加して水とする一酸素添加酵素(monooxygenase)とに区分される。日本の早石修ら[1][2][3]及び米国のHoward S. Masonら[4][5]の2つ研究グループから1955年の同時期に独立して発見が報告された。これらの違いはH.S.Masonらにより18O2の取り込みを研究されたことにより発見された(1955年)。早石は「酸素添加酵素群の発見と構造および生物学的意義の発見」にたいして、1986年のウルフ賞医学賞[6]を受賞している。
代表的な酸化還元酵素としてはチトクロムP450やモノフェノール酸化酵素などが知られている。
関連項目
· 酸化還元酵素
出典
· 酸素添加酵素『生化学辞典』第4版、岩波書店。
· オキシゲナーゼ『理化学辞典』第5版、岩波書店。
引用文献
1. ^ Hayaishi et al. (1955) Mechanism of the pyrocatechase reaction, J. Am. Chem. Soc. 77 (1955) 5450-5451
2. ^ Sligar SG, Makris TM, Denisov IG (2005). “Thirty years of microbial P450 monooxygenase research: peroxo-heme intermediates--the central bus station in heme oxygenase catalysis”. Biochem. Biophys. Res. Commun. 338 (1): 346–54. doi:10.1016/j.bbrc.2005.08.094. PMID 16139790.
3. ^ Hayaishi O (2005). “An odyssey with oxygen”. Biochem. Biophys. Res. Commun. 338 (1): 2–6. doi:10.1016/j.bbrc.2005.09.019. PMID 16185652.
4. ^ Mason HS, Fowlks WK, and Peterson E. (1955) Oxygen transfer and electron transport by the phenolase complex. J. Am. Chem. Soc.; 77(10) pp 2914 - 2915
5. ^ Waterman MR (2005). “Professor Howard Mason and oxygen activation”. Biochem. Biophys. Res. Commun. 338 (1): 7–11. doi:10.1016/j.bbrc.2005.08.120. PMID 16153596.
6. ^ “The Medicine Prize Committee unanimously decided that the Wolf Prize in Medicine for 1986 be awarded to Osamu Hayaishi”. Wolf Foundation. 2014年5月12日閲覧。
カテゴリ:
· 酸化還元酵素
酸化還元酵素酸化還元酵素に関するカテゴリ。EC番号ではEC 1に分類される。 下位カテゴリこのカテゴリには下位カテゴリ 22 件が含まれており、そのうち以下の22 件を表示しています。 E
カテゴリ「酸化還元酵素」にあるページこのカテゴリには 21 ページが含まれており、そのうち以下の 21 ページを表示しています。 *CPあおかさしたちとひふもれ |
DNAグリコシラーゼ
ウラシルDNAグリコシラーゼの構造。
DNAグリコシラーゼ(DNA glycosylase、EC 3.2.2.3)は、DNAのN-グリコシド結合を加水分解する酵素の総称で、塩基除去修復において傷害のある塩基をDNAから取り除く役割を担う。チミンDNAグルコシラーゼ英語版、ウラシルDNAグルコシラーゼ英語版、オキソグアニングルコシラーゼ英語版など、傷害塩基の種類によってさまざまなものが存在する。反応の結果生じた塩基の無い部位(AP site)は、APエンドヌクレアーゼ、DNAリガーゼ等の塩基除去修復経路の下流の酵素によって処理される。
ウラシルDNAグルコシラーゼは、PCR産物間のコンタミネーションの防止のためにも使われている[1]。この他、ヒトの解糖系での反応を触媒する酵素の1つで、4量体で活性を示すグリセルアルデヒド-3-リン酸デヒドロゲナーゼの単量体が、ヒトのウラシルDNAグルコシラーゼと同じ物であることが判明した[2]。
出典
2. ^ 『A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceroaldehyde-3-phospholate dehydrogenase.』
参考文献
· IUBMB entry for 3.2.2.3(英語)
· BRENDA references for 3.2.2.3 (英語)
· PubMed references for 3.2.2.3(英語)
· PubMed Central references for 3.2.2.3(英語)
· Google Scholar references for 3.2.2.3(英語)
関連項目
· 加水分解酵素
外部リンク
· IUBMB entry for 3.2.2.3(英語)
· KEGG entry for 3.2.2.3(英語)
· BRENDA entry for 3.2.2.3(英語)
· NiceZyme view of 3.2.2.3(英語)
· EC2PDB: PDB structures for 3.2.2.3(英語)
· PRIAM entry for 3.2.2.3(英語)
· PUMA2 entry for 3.2.2.3(英語)
· IntEnz: Integrated Enzyme entry for 3.2.2.3(英語)
· MetaCyc entry for 3.2.2.3(英語)
· Atomic-resolution structures of enzymes belonging to this class(英語)
か
解離定数
化学、生化学、薬理学において、解離定数(かいりていすう、![]() )は、複合体がその構成分子へとばらばらになる時、あるいは塩がその構成イオンへと分かれる時に、より大きな方の対象物がより小さな構成要素へと可逆的に分離(解離)する傾向を測る特殊な平衡定数である。解離定数は結合定数の逆数である。塩についての特別な場合は、解離定数はイオン化定数とも呼ばれる。
)は、複合体がその構成分子へとばらばらになる時、あるいは塩がその構成イオンへと分かれる時に、より大きな方の対象物がより小さな構成要素へと可逆的に分離(解離)する傾向を測る特殊な平衡定数である。解離定数は結合定数の逆数である。塩についての特別な場合は、解離定数はイオン化定数とも呼ばれる。
複合体![]() がx Aサブユニットとy Bサブユニットへと別れる一般的な反応
がx Aサブユニットとy Bサブユニットへと別れる一般的な反応
について、解離定数は以下のように定義される。
上式において、[A]、[B]、[AxBy] はそれぞれA、B、複合体AxByの濃度である。
生化学および薬理学において解離定数の人気がある一つの理由は、x=y=1となるしばしば見られる場合において、Kdが単純な物理学的解釈を有することである。[A]=Kdの時、[B]=[AB] あるいは [AB]/([B]+[AB])=1/2である。つまり、濃度の次元を有するKdは、Bの全分子の半数がAと会合している時の遊離のAの濃度に等しい。この単純な解釈はxあるいはyがより大きな値を取る場合には当てはまらない。また、競合反応が存在しないことも仮定されているが、競合的結合をあらわに扱い、記述できるように導出を拡張することができる。EC50やIC50が物質の生物学的活性を説明するのと同じように、解離定数は物質の結合の素早い説明として有用である。
目次
タンパク質-リガンド結合
解離定数はリガンド (![]() )
(薬剤など)とタンパク質 (
)
(薬剤など)とタンパク質 (![]() )
との間の親和性(すなわちリガンドが特定のタンパク質にどのぐらい強く結合しているか)を説明するために一般的に使われている。リガンド-タンパク質親和性は水素結合や静電相互作用、疎水性相互作用、ファンデルワールス力といった2分子間の非共有結合性相互作用によって影響を受ける。また、高濃度の他の高分子によっても影響を受け、分子クラウディングの原因となる[1][2]。
)
との間の親和性(すなわちリガンドが特定のタンパク質にどのぐらい強く結合しているか)を説明するために一般的に使われている。リガンド-タンパク質親和性は水素結合や静電相互作用、疎水性相互作用、ファンデルワールス力といった2分子間の非共有結合性相互作用によって影響を受ける。また、高濃度の他の高分子によっても影響を受け、分子クラウディングの原因となる[1][2]。
リガンド-タンパク質複合体(![]() )は2つの状態を含む過程によって記述できる。
)は2つの状態を含む過程によって記述できる。
対応する解離定数は以下のように定義される。
上式において、[![]() ]、[
]、[![]() ]、[
]、[![]() ]
は、それぞれタンパク質、リガンド、複合体のモル濃度を表わす。
]
は、それぞれタンパク質、リガンド、複合体のモル濃度を表わす。
解離定数はモル濃度単位 (M) を持ち、特定のタンパク質の結合部位の半分が占有されるリガンドの濃度 [![]() ]
に一致する。すなわち、リガンドが結合したタンパク質の濃度 [
]
に一致する。すなわち、リガンドが結合したタンパク質の濃度 [![]() ]
がリガンドが結合していないタンパク質の濃度 [
]
がリガンドが結合していないタンパク質の濃度 [![]() ]
と等しくなるリガンドの濃度である。解離定数が小さくなるとリガンドはよりしっかりと結合する、あるいはリガンドとタンパク質との間の親和性が高まる。例えば、ナノモーラー (nM) オーダーの解離定数を有するリガンドは、マイクロモーラー (
]
と等しくなるリガンドの濃度である。解離定数が小さくなるとリガンドはよりしっかりと結合する、あるいはリガンドとタンパク質との間の親和性が高まる。例えば、ナノモーラー (nM) オーダーの解離定数を有するリガンドは、マイクロモーラー (![]() M) オーダーの解離定数を有するリガンドよりも特定のタンパク質によりしっかりと結合する。
M) オーダーの解離定数を有するリガンドよりも特定のタンパク質によりしっかりと結合する。
2分子間の非共有結合性相互作用によって生じるピコモーラーより小さい解離定数は稀である。にもかかわらず、いくつかの重要な例外が存在する。ビオチンとアビジンは、おおよそ
![]() M = 1 fM = 0.000001 nMの解離定数で結合する[3]。
M = 1 fM = 0.000001 nMの解離定数で結合する[3]。
また、リボヌクレアーゼインヒビタータンパク質もリボヌクレアーゼと同じような![]() Mの親和性で結合できる[4]。特定のリガンド-タンパク質複合体に対する解離定数は溶液条件(例えば温度、pH、塩濃度)によって著しく変化する。異なる溶液条件の影響は、特定のリガンド-タンパク質複合体を結び付けている全ての非共有結合性相互作用の強さを効果的に変更する。
Mの親和性で結合できる[4]。特定のリガンド-タンパク質複合体に対する解離定数は溶液条件(例えば温度、pH、塩濃度)によって著しく変化する。異なる溶液条件の影響は、特定のリガンド-タンパク質複合体を結び付けている全ての非共有結合性相互作用の強さを効果的に変更する。
薬剤は、相互作用するように意図あるいは設計されていないタンパク質との相互作用によって有害な副作用を生じうる。ゆえに、たくさんの薬理学的研究が標的タンパク質のみに対して高い親和性(通常0.1-10 nM)で結合する薬剤の設計、あるいは特定の薬剤とそのin vivo標的タンパク質との間の親和性の向上を対象としている。
抗体
抗体 (Ab) が抗原 (Ag) に結合する特殊な場合においては、大抵は親和性定数が用いられる。これは解離定数の逆数である。
この化学平衡は、会合速度定数(kforward)と解離速度定数(kback)との比でもある。2つの抗体が同じ親和性を持つ場合もあるが、一方が高い会合速度定数と低い解離速度定数、他方が低い会合速度定数と高い解離速度定数を持つためかもしれない。
酸-塩基反応
詳細は「酸解離定数」を参照
酸の脱プロトン化に対するKは、酸解離定数Kaとして知られている。より強い酸、例えば硫酸あるいはリン酸がより大きな解離定数を持ち、酢酸のようなより弱い酸はより小さな解離定数を持つ。
酸解離定数は、 ![]() で定義されるp
で定義されるp![]() によって表わされることがある。
によって表わされることがある。
この![]() 表記は同様にその他の文脈でも見られる。共有結合の解離(すなわち化学結合の形成あるいは切断反応)では解離定数が非常に大きく変化するため、対数表記が主に用いられる。
表記は同様にその他の文脈でも見られる。共有結合の解離(すなわち化学結合の形成あるいは切断反応)では解離定数が非常に大きく変化するため、対数表記が主に用いられる。
分子は複数の酸解離定数を持ちうる。この点については、与えることができるプロトンの数に依存しており、一塩基酸、二塩基酸、三塩基酸を定義できる。一塩基酸(例えば酢酸やアンモニウム塩)は1つの解離性基のみを持ち、二塩基酸(炭酸、重炭酸塩、グリシン)は2つの解離性基、三塩基酸(例えばリン酸)は3つの解離性基を持つ。複数のpK値を持つ場合、それらはpK1やpK2、pK3といった指標によって指定される。アミノ酸では、pK1定数はカルボキシル基 (-COOH) を指し、pK2はアミノ基 (-NH3) を指し、pK3は側鎖のpK値である。
水の解離定数
詳細は「自己解離」を参照
水の解離定数はKwで示される。
水の濃度![]() は、慣習によって省略される。これはKwの値が濃度を用いて計算されたKeqの値とは異なることを意味する。
は、慣習によって省略される。これはKwの値が濃度を用いて計算されたKeqの値とは異なることを意味する。
Kwの値は、下記の表に示すように温度によって変化する。pHといった量の精密な測定を行う時にはこの変化を考慮に入れなければならない。
|
水の温度 |
Kw / 10−14 |
pKw[5] |
|
0℃ |
0.112 |
14.95 |
|
25℃ |
1.023 |
13.99 |
|
50℃ |
5.495 |
13.26 |
|
75℃ |
19.95 |
12.70 |
|
100℃ |
56.23 |
12.25 |
脚注
1. ^ Zhou HX, Rivas G, Minton AP (2008). “Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences”. Annu. Rev. Biophys. 37: 375-397. doi:10.1146/annurev.biophys.37.032807.125817. PMC 2826134. PMID 18573087.
2. ^ Minton AP. (2001). “The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media”. J. Biol. Chem. 276 (14): 10577-10580. doi:10.1074/jbc.R100005200. PMID 11279227.
3. ^ Livnah O, Bayer EA, Wilchek M, Sussman JL (1993). “Three-dimensional structures of avidin and the avidin-biotin complex”. Proc. Natl. Acad. Sci. U. S. A. 90 (11): 5076-5080. PMC 46657. PMID 8506353.
4. ^ Johnson RJ, McCoy JG, Bingman CA, Phillips GN Jr, Raines RT (2007). “Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein”. J. Mol. Biol. 368 (2): 434-449. doi:10.1016/j.jmb.2007.02.005. PMC 199390. PMID 17350650.
5. ^ Bandura, Andrei V.; Lvov, Serguei N. (2006). “The Ionization Constant of Water over Wide Ranges of Temperature and Density”. Journal of Physical and Chemical Reference Data 35 (1): 15-30. doi:10.1063/1.1928231.
関連項目
|
||
|
|
|
活性部位
この記事は検証可能な参考文献や出典が全く示されていないか、不十分です。
出典を追加して記事の信頼性向上にご協力ください。(2011年4月)
酵素反応の誘導適合モデル
分子生物学における活性部位(かっせいぶい、英: active site)は、基質が結合し化学反応が進む酵素の部位のことである。多くの酵素はタンパク質からできているが、リボザイムと呼ばれるリボ核酸でできた酵素も存在する。酵素の活性部位は、基質の認識に関わるアミノ酸(又は核酸)が並んだ溝又はポケットで見られる。触媒反応に直接関わる残基は、活性部位残基と呼ばれる。
1結合機構
2化学
3関連項目
4外部リンク
結合機構
酵素の作用機構には、「鍵と鍵穴モデル」と「誘導適合モデル」の2つのモデルが提案されている。鍵と鍵穴モデルは、活性部位は特定の基質と完全にぴったり合うものであり、一度基質が酵素に結合するとそれ以上の修正は必要ないという最も簡便なモデルである。誘導適合モデルは、鍵と鍵穴モデルを発展させたもので、活性部位はより柔軟であり、活性部位でのある残基の存在によって正しい基質が配置され、基質が結合した後に構造変化が起こると考えるものである。
化学[編集]
基質は水素結合、疎水結合、ファンデルワールス力等によって酵素の活性部位と結合する。活性部位の残基はプロトンや基質のその他の官能基のドナーやアクセプターとして働く。言い換えると、活性部位は反応の活性化エネルギーに応じて反応機構を変える。生成物は立体障害により活性部位では不安定となるために放出され、酵素は当初の非結合状態に戻る。
関連項目[編集]
酵素反応
ヒュー・テイラー
外部リンク[編集]
Catalytic Site Atlas (CSA) — hosted by EMBL-EBI
[隠す]
表 話 編 歴
タンパク質:酵素
トピックス
活性部位 アロステリック効果 結合部位 触媒三残基 補酵素 補因子 共同性 EC番号 酵素反応 酵素阻害剤 酵素反応速度論 ミカエリス・メンテン式
タイプ
EC1 酸化還元酵素 EC2 転移酵素 EC3 加水分解酵素 EC4 リアーゼ EC5 異性化酵素 EC6 リガーゼ
カテゴリ: 酵素
カテコールオキシダーゼ
|
カテコールオキシダーゼ |
||
|
識別子 |
||
|
データベース |
||
|
PDB構造 |
||
|
||
カテコールオキシダーゼ (catechol oxidase; EC 1.10.3.1) とは、酵素の一種で、チロシナーゼ、ジフェノールオキシダーゼ、カテゴラーゼ、o-ジフェノラーゼ、フェノラーゼ等の別名を持つ。カテコールオキシダーゼは以下の反応を触媒する。
2 カテコール + O2 ⇌ {\displaystyle
\rightleftharpoons } ![]() 2 1,2-benzoquinone + 2 H2O
2 1,2-benzoquinone + 2 H2O
|
|

また、チロシナーゼのような銅を含むカテコールオキシダーゼは、EC 1.14.18.1に分類されるモノフェノールモノオキシゲナーゼとしての活性も持っている。その反応のようすを以下に示す。
|
|

L-チロシン + L-ドーパ + O2
⇌
{\displaystyle \rightleftharpoons } ![]() L-ドーパ + ドーパキノン + H2O
L-ドーパ + ドーパキノン + H2O
なお、動物由来のものはチロシン、ドーパに対する活性が高いといわれている。
参考文献
· Solomon, E.I.; Chen, P.; Metz, M.; Lee, S.-K.; Palmer, A.E. (2001). “Oxygen Binding, Activation, and Reduction to Water by Copper Proteins”. Angew. Chem. Int. Ed. 40: 4570–4590. doi:10.1002/1521-3773(20011217)40:24<4570::AID-ANIE4570>3.0.CO;2-4. PMID 12404359.
β-ラクタマーゼ
β-ラクタマーゼ(ベータラクタマーゼ、β-lactamase)とはβ‐ラクタム系抗生物質を加水分解する酵素である。ペニシリン/セファロスポリンアミド-β-ラクタムヒドロラーゼ (penicillin/cepharosporin amido-β-lactam hydrolase)とも呼ばれる。EC3.5.2.6に分類される酵素である。
幾つかの種類のグラム陰性菌がβ-ラクタマーゼを産生することでβ-ラクタムに対して耐性を示すことが知られている。なお、β-ラクタム耐性はβ-ラクタマーゼのみが原因ではなくMRSAのようにペニシリン結合タンパク質の基質特異性が変化しても現れる。
現在β-ラクタマーゼは基質特異性の違いにより
· ペニシリナーゼ (クラスA β-ラクタマーゼ)
· メタロ-β-ラクタマーゼ (クラスB β-ラクタマーゼ、亜鉛-β-ラクタマーゼ、カルバペネマーゼ)
· セファロスポリナーゼ (クラスC β-ラクタマーゼ)
· オキサシリナーゼ (クラスD β-ラクタマーゼ)
これら4種のβ-ラクタマーゼのうち、クラスB β-ラクタマーゼは活性中心に亜鉛を持つが、他はセリン残基を持つ。ペニシリナーゼはペニシリン系抗生物質と第二世代セファロスポリンを分解するのに対して、セファロスポリナーゼは主にセファロスポリンを分解する。オキサシリナーゼはオキサシリンをも分解するペニシリナーゼであり、メタロ-β-ラクタマーゼはカルバペネム系抗生物質を分解する点に特徴がある。
β-ラクタマーゼの遺伝子は、細菌の染色体上あるいはプラスミド上に存在する。特に伝達性薬剤耐性プラスミド (drug resistance plasmid)に存在するβ-ラクタマーゼ遺伝子は菌種特異性も少なく多剤耐性菌の発生にも関与していると考えられる。
脚注
1. ^ β-ラクタム耐性菌とその検出方法、関東化学
2. ^ Bush, K. et. al. A functional classification scheme for β-lactamases and its correlation with molecular structure, Antimicrob Agents Chemother., 39, 1211-1233, 1995.
3. ^ Ambler, R. P., The structure of β-lactameses, Philos Trans R Society Lond (Biol), 289, 321-331, 1980.
4. ^ 石井良和、基質特異性拡張型β-ラクタマーゼ産生大腸菌、クレブシエラ、臨床と微生物、26、121-125, 1999.
関連項目
· クラブラン酸
· 薬剤耐性
· ペニシリン
· セファロスポリン
出典
· β-ラクタマーゼ『生物学辞典』第4版、岩波書店。
· β-ラクタマーゼについて 日本ベクトン・ディッキンソン株式会社
γ-グルタミルトランスフェラーゼ
|
γ-グルタミルトランスフェラーゼ |
|||||||||
|
識別子 |
|||||||||
|
データベース |
|||||||||
|
PDB構造 |
|||||||||
|
|||||||||
|
γ-グルタミルトランスフェラーゼ1 |
|
|
識別子 |
|
|
略号 |
|
|
遺伝子コード |
GGT |
|
他のデータ |
|
|
EC番号 |
|
|
γ-グルタミルトランスフェラーゼ2 |
|
|
識別子 |
|
|
略号 |
GGT2 |
|
遺伝子コード |
GGT |
|
他のデータ |
|
|
EC番号 |
|
γ-グルタミルトランスフェラーゼ(ガンマグルタミルトランスフェラーゼ、γ-glutamyltransferase; γ-GT, GGT; EC 2.3.2.2)は、グルタチオンなどのγ-グルタミルペプチドを加水分解し、他のペプチドやアミノ酸にγ-グルタミル基を転移する酵素。γ-グルタミルトランスペプチターゼ(γ-glutamyl transpeptidase; γ-GTP, GGTP)とも呼ばれる。
生体内ではそのほとんどが膜結合型酵素として存在し、膜を介したアミノ酸の移動に関与している。ヒトでは腎臓で最も活性が高く、さらに膵臓、肝臓、脾臓、小腸、精巣、前立腺など広く全身に分布する。
肝臓では、肝細胞のミクロソーム分画で産生され、細胆管、毛細胆管などの細胞膜に移動して機能している。これが閉塞性黄疸、肝癌、アルコール性肝障害など肝・胆道系の疾患で誘導され、逸脱酵素として血中に流出する。このため、血中のγ-グルタミルトランスフェラーゼ活性は肝機能の指標として利用されている。
関連項目
外部リンク
- MedlinePlus Encyclopedia 003458
- gamma-Glutamyltransferase - MeSH、米国国立医学図書館、生命科学用語シソーラス (英語サイト)
き
基質 (化学)
基質(英語:substrate)とは、化学反応において他の試薬と反応して生成物を作る化学種の1つである。合成化学や有機化学においては、基質の化合物にわずかに修正を加えて目的の物質へと変換する。 生化学においては酵素と結合して酵素が働く場所となる物質を基質と呼ぶ。ルシャトリエの原理より、基質は濃度が変化する物質である。「基質」という言葉が指すものは文脈によって大きく異なる[1]。
自発的反応
S → P {\displaystyle S\rightarrow P}
Sが基質、Pが生成物(英語版)
触媒反応
S + C → P + C {\displaystyle S+C\rightarrow P+C}
Sが基質、Pが生成物、Cが触媒。
生化学[編集]
生化学では、基質とは酵素と結合し、酵素が働く元となる分子である。酵素は基質が関わる化学反応を触媒する。
関連項目
|
化学反応 |
試薬 |
触媒 |
酵素 |
生成物 |
溶媒 |
脚注
キナーゼ
キナーゼ(Kinase、読み:カイネイス、カイネース)とは、生化学において、ATPなどの高エネルギーリン酸結合を有する分子からリン酸基を基質あるいはターゲット分子に転移する(リン酸化する)酵素の総称であり、リン酸化酵素とも呼ばれる。EC 2.7群(リン酸転移酵素、ホスホトランスフェラーゼ)に属する。英語発音に由来するカイネイス、カイネースと呼ぶ研究者が増えてきている。
一般に高エネルギーリン酸化合物からのリン酸転移反応は大きな負の自由エネルギー変化を伴うため不可逆変化として進行しやすく、その結果生じる化合物もまた高エネルギーリン酸化合物である場合もある。ゆえにキナーゼは基質分子に対して「活性化」あるいは「エネルギーを与える」(キナーゼの名称もこの意味による)と考えることができる。すべてのキナーゼはMg2+あるいはMn2+など2価の金属イオンを要し、それによりドナー分子の末端リン酸基の転移を容易にする。
キナーゼには様々なタイプがあるが、大きくは低分子化合物を基質とし代謝経路で機能するタイプと、タンパク質を基質としてその機能を調節したり細胞内シグナル伝達経路で機能するタイプの2つに分けられる。例として次のようなものがある:
低分子基質タイプ
クレアチンキナーゼ
ピルビン酸キナーゼ
ヘキソキナーゼ
プロテインキナーゼ:特にこのタイプをキナーゼとよぶことも多い。
なおプロテアーゼの中にはウロキナーゼ、ナットウキナーゼのように「キナーゼ」という名称のついたものがあるが、これらは「酵素(プラスミノーゲン)を活性化する酵素」の意味で個別に命名されたものであって、本項目のキナーゼではない。
[隠す]
表 話 編 歴
ホスホトランスフェラーゼ/キナーゼ (EC 2.7)
2.7.1 - OH アクセプター
ヘキソ- - グルコ- - フルクト- - ガラクト- - ホスホフルクト- - チミジン - NAD+- - グリセロール- - パントテン酸- - メバロン酸- - ピルビン酸- - デオキシシチジン- - PFP - ジアシルグリセロール- - ブルトンチロシン - ホスホイノシチド-3 - スフィンゴシン
2.7.2 - COOH アクセプター
ホスホグリセリン酸 - アスパラギン酸
2.7.3 - N アクセプター
クレアチン
2.7.4 - PO4 アクセプター
ホスホメバロン酸 - アデニル酸 - ヌクレオシド二リン酸
2.7.6 - P2O7トランスフェラーゼ
リボースリン酸ジホスホキナーゼ - チアミンピロホスホキナーゼ
2.7.7 - ヌクレオチジル-
インテグラーゼ - PNPアーゼ - ポリメラーゼ - RNアーゼ PH - UDP-グルコースピロホスホリラーゼ - ガラクトース-1-リン酸ウリジリルトランスフェラーゼ -ターミナルトランスフェラーゼ - RNAレプリカーゼ - リバーストランスクリプターゼ (テロメラーゼ) - トランスポザーゼ
2.7.8 - 他のリン酸基
N-アセチルグルコサミン-1-リン酸トランスフェラーゼ
2.7.10-11 - プロテイン
チロシン - セリン/トレオニンプロテイン
金属タンパク質
金属タンパク質(きんぞくタンパクしつ、Metalloprotein)は、補因子として金属を含むタンパク質を表す用語である。金属は単独のイオンかあるいはタンパク質以外のポルフィリンなどの有機化合物に配位して存在している。タンパク質の側鎖や非金属無機イオンに配位している場合もある。このようなタンパク質-金属-非金属の構造は鉄-硫黄クラスターなどでも見られる。
金属タンパク質の内重要なものに金属酵素がある。これは、その活性中心の中に1つか2つの金属原子を含むものである。このような金属は、炭酸脱水酵素やシトクロムcオキシダーゼの場合のように触媒活性に関わっていることもしばしばある。金属イオンは通常複数の配位をして活性部位の一部となり、孤立電子対によって基質との高い親和性を作っている。
金属タンパク質の例
|
イオン |
イオンを含む酵素の例 |
銅 |
シトクロムcオキシダーゼ |
鉄 |
|
カタラーゼ |
シトクロム(ヘム) |
ニトロゲナーゼ |
ヒドロゲナーゼ |
マグネシウム |
|
グルコース 6-ホスファターゼ |
ヘキソキナーゼ |
マンガン |
アルギナーゼ |
モリブデン |
|
硝酸還元酵素 |
ニッケル |
ウレアーゼ |
セレン |
グルタチオンペルオキシダーゼ |
|
亜鉛 |
アルコールデヒドロゲナーゼ |
炭酸脱水酵素 |
DNAポリメラーゼ |
|
関連項目
|
補酵素 |
補因子 |
生物無機化学 |
補欠分子族 |
ヘムタンパク質 |
銅タンパク質 |
く
クエン酸シンターゼ
|
Citrate synthase |
|
|
識別子 |
|
|
略号 |
CS |
|
他のデータ |
|
|
EC番号 |
|
クエン酸シンターゼ(クエンさんシンターゼ、Citrate synthase)は、ほぼ全ての生細胞に含まれ、クエン酸回路の第一段階の速度を調整する酵素である[1]。クエン酸シンターゼは、真核生物細胞のミトコンドリアマトリックスに局在するが、ミトコンドリアではなく細胞核のDNAによってコードされる。細胞質のリボソームで合成され、その後ミトコンドリアのマトリックスに輸送される。クエン酸シンターゼは、完全なミトコンドリアの存在量を示すマーカーとしても用いられている。
クエン酸シンターゼは、アセチルCoAの酢酸残基をオキサロ酢酸に付加し、クエン酸を合成する反応を触媒する[1]。オキサロ酢酸は、クエン酸回路を一周すると再生される。
|
|
|
|



アセチルCoA + オキサロ酢酸 + 水 → クエン酸 + 補酵素A
オキサロ酢酸が最初に酵素に結合すると、酵素の形が変化し、アセチルCoAの結合部位が形成される。シトロイルCoAが生成するとさらに構造が変化し、チオエステルを加水分解し、補酵素Aを遊離する。これにより、チオエステル結合の切断により放出されるエネルギーが縮合反応を駆動する。
構造
|
|
|
|
クエン酸シンターゼ(閉状態)の活性部位 |
クエン酸シンターゼ(開状態)の活性部位 |

.png)
クエン酸シンターゼの437個の残基は、それぞれ20個のαヘリックスを持つ2つのサブユニットを構成する。
αヘリックスは、クエン酸シンターゼの構造の約75%を占め、その他は13残基のβシートとランダムな構造である。この2つのサブユニットの間の溝に活性部位が存在する。その中に2つの結合部位があり、そのうち1つはクエン酸またはオキサロ酢酸、もう1つは補酵素Aを保持するものである。活性部位には、3つの重要な残基His274、His320、Asp375が存在し、基質への作用の選択性は高い。
左図は、クエン酸シンターゼの三次構造である。酵素は、どちらかの基質が結合することで上図(開状態)から下図(閉状態)に構造が変化する[2]。
機構
クエン酸シンターゼは、アセチルCoA(H3CCO-SCoA)とオキサロ酢酸(COO-CH2COCOO-) からクエン酸(COO-CH2COHCOOCH2COO-)と補酵素A (H-SCoA) への変換を触媒する活性中心に、3つの重要なアミノ酸残基を持つ。この化学変化は、Asp375の側鎖の負電荷を持った酸素原子がアセチルCoAのα炭素を脱プロトン化することで開始する。これにより電子が押し出され、カルボニル基の炭素原子と二重結合を形成し、His274の側鎖のどちらかの窒素原子からC=Oの酸素原子にプロトンを与える。これにより、窒素原子上に不対電子が形成されて側鎖が中和され、エノール中間体 (CH2COH-SCoA) が生成する。この時点で、His274のアミノ基の不対電子が酸素原子に結合したプロトンを攻撃する。酸素原子は、カルボニル結合を回復し、C=C二重結合を単結合にして、オキサロ酢酸のカルボニル炭素 (COO-CH2COCOO-) を求核攻撃させる。これにより、カルボニル結合の二重結合が単結合になり、His320のアミノ基の1つを脱プロトン化し、側鎖の窒素原子の1つが中和される。この求核付加反応により、シトロイルCoA (COOCH2CHCOOCH2COHSCoA2-) が生成する。この時点で、水分子が導入され、His320のアミノ基によって脱プロトン化されて加水分解が始まる。酸素原子の1つの不対電子がシトロイルCoAのカルボニル炭素を求核攻撃し、四面体型の中間体を形成し、-SCoA基が外れてカルボニルが再形成される。-SCoA基はプロトン化されて補酵素A (HSCoA) となる。最終的に、前の段階でカルボニル基に付加したヒドロキシル基が脱プロトン化し、クエン酸 (-COOCH2COHCOO-CH2COO-) が形成される[3]。

クエン酸シンターゼの活性機構
阻害
高濃度のATP、アセチルCoA、NADHが存在すると、エネルギー供給が細胞にとって高すぎるため、この酵素は、ATP:ADP、アセチルCoA:CoA、NADH:NADが高い比率になると阻害される。また、スクシニルCoAやクエン酸によって生成物阻害が起こる。アセチルCoAのアナログによるクエン酸シンターゼの阻害は、良く研究されており、単一の活性中心が存在すると証明するために重要な役割を果たした。これらの実験により、この活性中心が2つの形をとり、それぞれがリガーゼとヒドロラーゼの活性に関与することを明らかとした[4]。このタンパク質は、アロステリック制御のモデルとしても用いられる[5]。
経路図
以下の遺伝子、タンパク質、代謝それぞれの記事をクリックできる [6]
[[File:

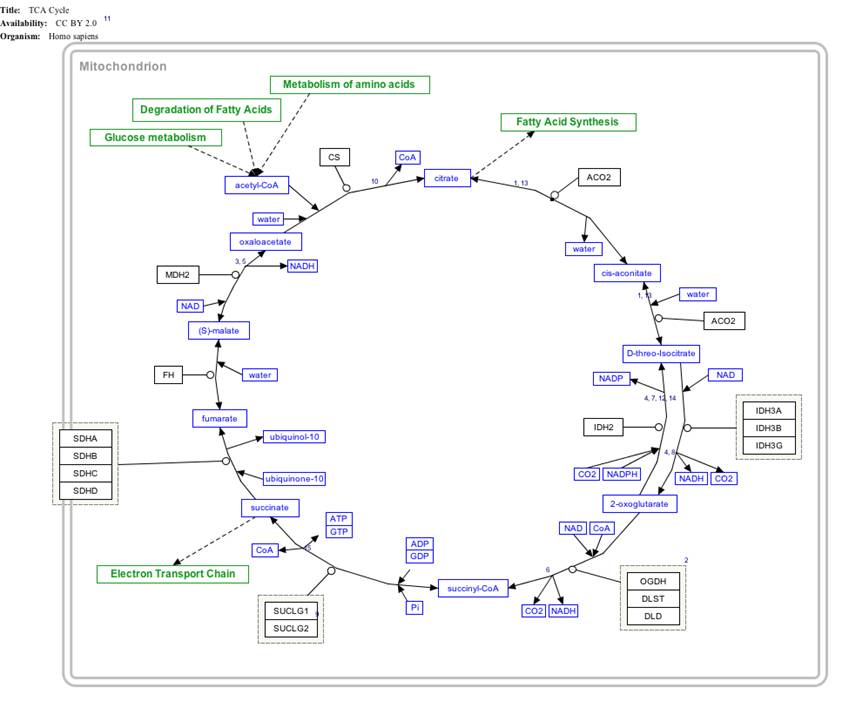
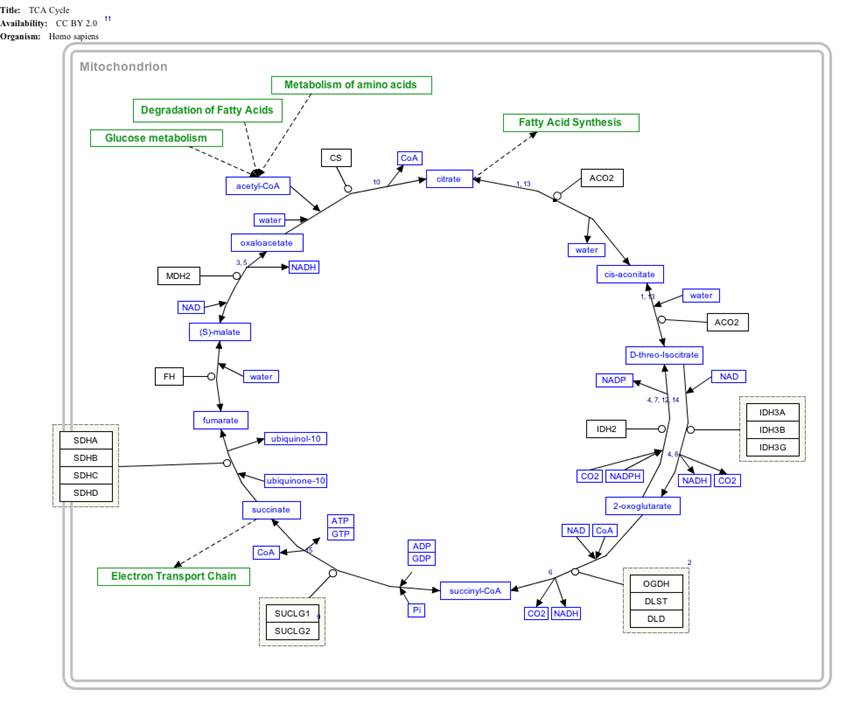
|{{{bSize}}}px|class=noresize]]
クエン酸回路 編集
出典
- ^ a b Weigand, Georg, and Steven J. Remington (1986). “Citrate Synthase: Structure, Control, and Mechanism”. Ann. Rev. Biophys. Biophys. Chem.: 98. doi:10.1146/annurev.bb.15.060186.000525.
- ^ Ernat BAYER, Barbara BAUER, Hermann EGGERER (1981). “Evidence from Inhibitor Studies for Conformational Changes of Citrate Synthase”. Eur J Biochem 120: 155-160. doi:10.1111/j.1432-1033.1981.tb05683.x.
- ^ Lehninger (2005). Principles of Biochemistry: Fourth Edition. W.H. Freeman and Co. Pages 608-609.
- ^ Ernat BAYER, Barbara BAUER, Hermann EGGERER (1981). “Evidence from Inhibitor Studies for Conformational Changes of Citrate Synthase”. Eur J Biochem 120: 155-160. doi:10.1111/j.1432-1033.1981.tb05683.x.
- ^ T. Selwood and E. K. Jaffe. (2011). “Dynamic dissociating homo-oligomers and the control of protein function.”. Arch. Biochem. Biophys. 519 (2): 131-43. doi:10.1016/j.abb.2011.11.020. PMC 3298769. PMID 22182754.
- ^ この双方向伝達経路地図はWikiPathwaysで編集できる: TCA_Cycle_WP78
外部リンク
- Citrate synthase - MeSH、米国国立医学図書館、生命科学用語シソーラス (英語サイト)
|
||||||
<img src="//ja.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1x1" alt="" title="" width="1" height="1" style="border: none; position: absolute;" />
「https://ja.wikipedia.org/w/index.php?title=クエン酸シンターゼ&oldid=66197694」から取得
カテゴリ:
グリコシダーゼ
|
|

グリコシダーゼの一種、α-アミラーゼ 1HNY の構造
グリコシダーゼ(glycosidase)とは、グリコシド結合を加水分解する酵素の総称であり、グリコシドヒドロラーゼ(glycoside hydrolase)とも呼ばれる。
主な役割として、バイオマスにおけるセルロースやヘミセルロースの分解、バクテリアに対する防御(例:リゾチーム)、ウイルスによる細胞への感染(例:ノイラミニダーゼ)、細胞内における糖タンパク質の生合成などに関係している。
グリコシダーゼは、グリコシド結合の形成や分解においてグリコシルトランスフェラーゼとともに重要な役割を担っている。

機能
グリコシダーゼは基本的に全てのドメインの生物に存在する。細胞内外の両方に存在し、栄養の吸収に関係している。バクテリアにおける最も重要なグリコシダーゼの1つにβ-ガラクトシダーゼ(LacZ)があり、大腸菌のラクトースオペロンの発現を調節している。
ゴルジ体や小胞体に存在するグリコシダーゼはN結合型糖タンパク質糖鎖のプロセシングに、リソソームに存在するものはさまざまな糖質の分解に関係している。リソソームの特定のグリコシダーゼが欠乏すると、本来分解されるはずの糖質が蓄積し、発達障害を起こしたり死亡したりする可能性がある(ライソゾーム病)。
消化管や唾液に存在するグリコシダーゼは、ラクトース、デンプン、スクロース、トレハロースといった糖を分解する働きがある。消化管内では、内皮細胞に定着したグリコシルホスファチジル(glycosylphosphatidyl)アンカー型酵素として存在している。
ラクターゼはラクトースの分解に必要な酵素であり、幼児期では高濃度に存在する。離乳後から濃度が減少し始め、成人期には量がかなり少なくなり、乳糖不耐症となる場合がある。
O-GlcNAcアーゼ(O-GlcNAcase)は細胞質や核内において、タンパク質のセリンやトレオニン残基からN-アセチルグルコサミンを取り除く働きをしている。
グリコシダーゼは体内でグリコーゲンの生合成と分解にも関係している。
分類
グリコシダーゼはEC 3.2.1 に分類され、O- または S-グリコシドの加水分解を触媒する。
加水分解の立体化学に基づいて、保持型(retaining)もしくは反転型(inverting)に分類することも出来る[1]。
活性がエキソ型(exo)かエンド型(endo)かによっても分類することが出来る。エキソ型はオリゴ糖や多糖の末端に作用し、エンド型は中間部に作用する。
また、シークエンスや立体構造に基づいて分類する方法もある。
シークエンスに基づいた分類
シークエンスに基づいた分類は、新しく配列が決定された酵素のまだ知られていない機能を予測するのに非常に重要な手段である。グリコシダーゼをシークエンスに基づいて分類した場合、100を超えるファミリーに分類することが出来る[2][3][4]。この分類は、CAZy(CArbohydrate-Active EnZymes)のウェブサイトにて利用することができる[5]。データベースは、シークエンス、予測されるメカニズム(保持型/転化型)、活性サイトの残基、基質に関する情報を提供している。
三次元構造の類似性に基づき、シークエンスで分類された「ファミリー」はクラン(clan)と呼ばれる上位カテゴリーでさらに分類される。シークエンス分析と三次元構造分析により、従来より幅広く分類できるようになった[6]。
メカニズム
反転型グリコシダーゼ
反転型グリコシダーゼ(inverting glycosidase)は、2つの残基(典型的にはカルボン酸、つまりアスパラギン酸残基かグルタミン酸残基)が1組となって触媒を行う。下の例はβ-グルコシダーゼのものであり、2つの触媒残基がそれぞれ酸と塩基の働きをする。

保持型グリコシダーゼ
保持型グリコシダーゼ(retaining glycosidase)は、2つのステップで反応を触媒する。それぞれのステップで立体構造をワルデン反転させるため、結果的にもとの構造を保持する。
反応には2つの残基が関係する。この残基は通常、酵素の有するカルボン酸基である。1つは求核剤、もう1つは酸/塩基として働く。
最初のステップで求核触媒残基はアノマー中心部にアタックし、中間体を生成する。この時、酸/塩基触媒残基はプロトンを供与する酸の役割を果たす。
次のステップでは、プロトンを奪われた酸/塩基触媒残基が塩基の働きをし、求核剤である水が中間体を加水分解する反応を助ける。最終的に加水分解された産物が生成される。下の例はニワトリ卵白のリゾチームである。[7]

保持型グリコシダーゼにはもう1つの触媒メカニズムがある。この場合は求核残基が酵素ではなく、基質に結合することによって反応が進行する。このような反応は特定のN-アセチルヘキソサミニダーゼ(N-acetylhexosaminidase)で見られる。この酵素は隣接基と反応できるアセトアミド基を持っており、中間体としてオキサゾリン、もしくはオキサゾリニウムイオン(oxazolinium ion)を生成する。この場合もそれぞれのステップでワルデン反転を行うため、最終的に構造が保持される。

命名法
グリコシダーゼは通常、作用する基質に基づいて命名される。例えば、グルコシド結合を加水分解する酵素はグルコシダーゼ、キシロースのホモポリマーであるキシランを分解する酵素はキシラナーゼ(xylanase)と呼ばれる。
他にも、ラクターゼ、アミラーゼ、キチナーゼ、スクラーゼ、マルターゼ、ノイラミニダーゼ、インベルターゼ、ヒアルロニダーゼ、リゾチームなどがある。
用途
産業
グリコシダーゼは様々な分野で活用されている。
- 植物材料の分解
セルラーゼ、β-グルコシダーゼはセルロースをグルコースへ分解し、エタノールの生産に使用される。
- 食品産業
インベルターゼは転化糖の製造、アミラーゼはマルトデキストリンの製造に使用される。
- 製紙・パルプ産業
キシラナーゼはパルプからのヘミセルロースの除去に使用される。
- 洗剤
セルラーゼは衣服の色の鮮明化と衣服表面のマイクロファイバーの除去に使用される。
有機化学
平衡が逆向きの場合に加水分解の逆反応を行わせることで、グリコシド結合を付加させる触媒としても使用することができる。保持型グリコシダーゼは、活性化グリコシドからグリコシドを受け取る余裕のあるアルコールにグリコシル基の一部を転移することが出来る。
グリコシンターゼ(glycosynthase)はグリコシダーゼの変異体であり、フッ化糖のような活性化されたグルコシルドナーを利用し、高収率でグリコシドを合成できる。
阻害物質
グリコシダーゼの阻害物質は多くのものが知られている。デオキシノジリマイシン(deoxynojirimycin)、スワインソニン(swainsonine)、オーストラリン(australine)、カスタノスペルミン(castanospermine)のような、いくつかの窒素原子を含む糖型の複素環化合物が自然界から発見された。これらの天然由来の化合物から、イソファゴミン(isofagomine)、デオキシガラクトノジリマイシン(deoxygalactonojirimycin)、その他いくつかの不飽和化合物(例:PUGNAc)が阻害剤として開発された。
医薬品として使用されているグリコシダーゼ阻害剤には、アカルボース、ザナミビル(リレンザ)、ミグリトール(miglitol)、オセルタミビル(タミフル)などがある。
いくつかのタンパク質もグリコシダーゼを阻害することが判明した。
参考
1. ^ Sinnott, M. L. Chem. Rev. 1990, 90, 1171-1202.
2. ^ Henrissat B, Callebaut I, Mornon JP, Fabrega S, Lehn P, Davies G (1995). “Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases”. Proc. Natl. Acad. Sci. U.S.A. 92 (15): 7090-7094. PMID 7624375.
3. ^ Henrissat B, Davies G (1995). “Structures and mechanisms of glycosyl hydrolases”. Structure 3 (9): 853-859. PMID 8535779.
4. ^ Bairoch A (1999). Classification of glycosyl hydrolase families and index of glycosyl hydrolase entries in SWISS-PROT. pp. -.
5. ^ Henrissat B, Coutinho PM (1999). Carbohydrate-Active Enzymes server. pp. -.
6. ^ Naumoff, D.G. Proceedings of the Fifth International Conference on Bioinformatics of Genome Regulation and Structure. 2006, 1, 294-298.
7. ^ Vocadlo, D. J.; Davies, G. J.; Laine, R.; Withers, S. G. Nature 2001, 412, 835.
関連項目
外部リンク
グリシン開裂系
グリシン開裂系(Glycine cleavage system)は、グリシンデカルボキシラーゼ複合体(GCS)として知られている。グリシン開裂系は、アミノ酸である高濃度のグリシンに応答して活動を開始する一連の酵素である[1]。グリシン開裂系はT-タンパク質、P-タンパク質、L-タンパク質及びH-タンパク質の4種類のタンパク質で構成されている。これらのタンパク質は安定した複合体を形成しているわけではなく[2]、むしろ複合体と呼ぶより複雑なシステムと呼ぶ方が適切である。
グリシン開裂系の酵素の欠損は、ヒトに高グリシン血症をもたらす[3]。
構成
|
名前 |
機能 |
|
|
T-タンパク質 |
||
|
P-タンパク質 |
||
|
L-タンパク質 |
||
|
H-タンパク質 |
H-タンパク質 (GCSH) は、リポ酸で修飾され、(P-タンパク質によって触媒される)還元メチルアミノ化、(T-タンパク質によって触媒される)メチルアミン移転、(L-タンパク質によって触媒される)電子伝達のサイクルで他のすべてのコンポーネントと相互作用している[2]。 |
機能
グリシン開裂系はテトラヒドロ葉酸により以下の反応でグリシンを開裂する[4][信頼性要検証]。
テトラヒドロ葉酸 + グリシン + NAD+ = 5,10-メチレンテトラヒドロ葉酸+ NH3 + CO2 + NADH + H+
グリシン開裂系とは別に、グリシンヒドロキシメチルトランスフェラーゼ(セリンヒドロキシメチルトランスフェラーゼ)(EC 2.1.2.1)の働きにより、可逆的にグリシンをL-セリンに相互に変換し、5,10-メチレンテトラヒドロ葉酸をテトラヒドロ葉酸に変換する反応が触媒される[5][6]。
5,10-メチレンテトラヒドロ葉酸+ グリシン + H2O = テトラヒドロ葉酸 + L-セリン [7]
グリシン開裂系とセリンヒドロキシメチルトランスフェラーゼによる2つの反応を複合すると以下の反応式が示めされる。また、その全容は図の通りである。
2 グリシン + NAD+ + H2O → セリン + CO2 + NH3 + NADH + H+
グリシン開裂系によって拡張されたこの反応は、C3植物の光呼吸に必要である。
|

光呼吸経路におけるグリシン開裂、赤色の分子がグリシン。1はテトラヒドロ葉酸、2は5,10-メチレンテトラヒドロ葉酸、PLPはピリドキサールリン酸、TはT-プロテイン、PはP-プロテイン、LはL-プロテイン、HはH-プロテイン
葉酸との関係
テトラヒドロ葉酸(THF)による代謝とビタミンB12によるTHFの再生産、de:Folsäure=葉酸、DHF=ジヒドロ葉酸、THF=テトラヒドロ葉酸、Vit.B12=ビタミンB12、Methyl-Vit.B12=メチルコバラミン、Methionin=メチオニン、Methionin Syntase=5-メチルテトラヒドロ葉酸-ホモシステインメチルトランスフェラーゼ、Homocy
|
|

stein=ホモシステイン、N5-Methyl-THF=5-メチルテトラヒドロ葉酸、N5,N10-Methylene-THF=5,10-メチレンテトラヒドロ葉酸、N10-Formyl-THF=10-ホルミルテトラヒドロ葉酸、dUMP=デオキシウリジン一リン酸、NADPH、DNA
グリシン開裂系は神経幹細胞に存在することが明らかになっている。グリシン開裂系はDNA合成に必須である5,10-メチレンテトラヒドロ葉酸を供給している。グリシン開裂系酵素の欠損による非ケトーシス型高グリシン血症において、小頭症などの脳形成異常を高率に合併する機序として、5,10-メチレンテトラヒドロ葉酸の産生低下により神経幹細胞の増殖が障害される可能性が示唆されている。葉酸の触媒過程の全容において、平衡がテトラヒドロ葉酸側に偏っており、テトラヒドロ葉酸から5,10-メチレンテトラヒドロ葉酸を供給するグリシン開裂系は葉酸系の代謝過程で重要な役割を果たしている。
脚注
- ^ Kikuchi G (June 1973). “The glycine cleavage system: composition, reaction mechanism, and physiological significance”. Mol. Cell. Biochem. 1 (2): 169–87. doi:10.1007/BF01659328. PMID 4585091.
- ^ a b Douce R, Bourguignon J, Neuburger M, Rébeillé F (April 2001). “The glycine decarboxylase system: a fascinating complex”. Trends Plant Sci. 6 (4): 167–76. doi:10.1016/S1360-1385(01)01892-1. PMID 11286922.
- ^ 高グリシン血症 (難病情報センター)
- ^ アミノ酸の分解 講義資料のページ
- ^ Appaji Rao N, Ambili M, Jala VR, Subramanya HS, Savithri HS (April 2003). “Structure-function relationship in serine hydroxymethyltransferase”. Biochim. Biophys. Acta 1647 (1-2): 24–9. PMID 12686103.
- ^ Stover P, Schirch V (August 1990). “Serine hydroxymethyltransferase catalyzes the hydrolysis of 5,10-methenyltetrahydrofolate to 5-formyltetrahydrofolate”. J. Biol. Chem. 265 (24): 14227–33. PMID 2201683.
- ^ ENZYME: 2.1.2.1 KEGG
関連項目
アミラーゼ
アミラーゼ (amylase)とはジ(ヂ)アスターゼとも称される、膵液や唾液に含まれる消化酵素。グリコシド結合を加水分解することでデンプン(ラテン語"amylum")中のアミロースやアミロペクチンを、単糖類であるブドウ糖や二糖類であるマルトースおよびオリゴ糖に変換する酵素群である。
概要
アミラーゼは1833年、フランスの生化学者、アンセルム・ペイアン (Anselme Payen) とジャン・ペルソー (Jean F. Persoz) が大麦の芽から取り出し、「切り離す」を意味するギリシア語の “διαστασις” より「ジ(ヂ)アスターゼ」と命名された。これが酵素の初めての単離である。
アミラーゼは消化酵素であり、デンプンやグリコーゲンを分解する。体内では主に、膵臓、耳下腺(唾液腺)から分泌され、またダイコンやカブ、ヤマイモにも多く含まれている。胃腸薬、消化剤として市販もされ、胃もたれや胸焼けの治療、防止に服用されている。
日本の製薬会社三共の事実上の創業者である高峰譲吉は、麹菌からジアスターゼを抽出し、自身の名の「タカ」を冠してタカジアスターゼと命名して1894年(明治27年)に特許を申請した[1]。高峰のジアスターゼ(アミラーゼ)の抽出成功は古くから餅を食べるとき大根おろしをつけて食べると胃がもたれないと言う事が大きなヒントとなったとも伝えられる。
夏目漱石の作品『吾輩は猫である』には、佐伯矩が発見した大根ジアスターゼについてと思われる新聞記事やタカジアスターゼを常用する人物が描写されて[2]、消化を促進するという機能が広く知られ用いられた様子がわかる。[3]。
現在、正式な物質名はアミラーゼであるが、旧名であるジアスターゼも医薬品/化学薬品の『タカジアスターゼ』として使用されている。
日本では現在も第一三共の医療用医薬品として「タカヂアスターゼ末」として薬局に卸されている(主に解熱鎮痛剤や整腸剤など他の散剤と混合して使うが、処方箋医薬品ではないため零売が可能)。また、第一三共ヘルスケアから一般用医薬品(胃腸薬)の「新タカヂア錠」と「第一三共胃腸薬」シリーズにタカヂアスターゼNとして配合されている。アメリカではパーク・デイビス(現:ファイザー)から市販されていない。
異性体
α-アミラーゼ[4]、β-アミラーゼ[5]、グルコアミラーゼ[6]やイソアミラーゼ[7]がある。
α-アミラーゼ
|
α-アミラーゼ |
|
|
α-アミラーゼの構造 |
|
|
識別子 |
|
|
データベース |
|
|
PDB構造 |
|
α-アミラーゼは別名を1,4-α-D-グルカングルカノヒドロラーゼ、グリコゲナーゼといい、デンプンやグリコーゲンのα-1,4-結合を不規則に切断し、多糖ないしマルトース、オリゴ糖を生み出す酵素である。
β-アミラーゼ
|
β-アミラーゼ |
|
|
β-アミラーゼの構造 |
|
|
識別子 |
|
|
データベース |
|
|
PDB構造 |
|
β-アミラーゼは別名を1,4-α-D-グルカングルカノマルトヒドロラーゼ、グリコゲナーゼあるいはサッカロゲンアミラーゼといい、デンプンやグリコーゲンをマルトース(麦芽糖)に分解する。植物や微生物ではよく見られるが、動物からは見つかっていない。糖鎖の非還元末端から二つ目のα-1,4-グリコシド結合をエキソ型で逐次分解してマルトースを産生する。直鎖型のアミロースに対する分解効率は高い。一方、アミロペクチンに対してはα-1,6-グリコシド結合をしている分枝部で反応が停止し、マルトースとともにβリミットデキストリンが生成される。
グルコアミラーゼ
|
グルコアミラーゼ |
|
|
識別子 |
|
|
データベース |
|
|
PDB構造 |
|
グルコアミラーゼは正式名称がグルカン1,4-α-グルコシダーゼといい、1,4-α-D-グルカングルコヒドロラーゼ、エキソ1,4-α-グルコシダーゼ、γ-アミラーゼ、リソソーマルα-グルコシダーゼあるいはアミログルコシダーゼを別名とする。糖鎖の非還元末端のα-1,4-結合をエキソ型に加水分解してブドウ糖1分子を産生する。α-1,6-結合も切断するものも知られている。
イソアミラーゼ
詳細は「イソアミラーゼ」を参照
イソアミラーゼはアミロペクチンやグリコーゲン中のα-1,6-グリコシド結合を切断して直鎖のデキストリンやアミロースを生産する。ただし、プルランを分解できない。分枝部を切断するため、枝切り酵素や脱分枝酵素とも呼ばれる。
利用
アミラーゼは、植物では果実の成熟や穀物の発芽の間に合成される。穀物酒や酢、水あめなどの伝統的な製法ではデンプンの糖化に麦芽に含まれるアミラーゼが用いられる。
微生物の分泌するアミラーゼは工業的に大量に生産され、製糖、食品加工、胃腸薬、衣料製造、洗剤等に利用されている。工業的にアミラーゼを生産する微生物としてはアスペルギルス・オリゼーや枯草菌が知られている。
尿中や血中のアミラーゼは、膵臓疾患や唾液腺疾患の診断に使われる。
ヒトアミラーゼ
ヒトのアミラーゼには以下のものがある。
|
酵素名 |
遺伝子 |
遺伝子座標 |
機能 |
|
アルファ1A アミラーゼ(唾液腺) |
AMY1A,EC 3.2.1.1 |
1 p21 |
澱粉の分解 |
|
アルファ1B アミラーゼ(唾液腺) |
AMY1B,EC 3.2.1.1 |
1 p21 |
澱粉の分解 |
|
アルファ1C アミラーゼ(唾液腺) |
AMY1C,EC 3.2.1.1 |
1 p21 |
澱粉の分解 |
|
アルファ2A アミラーゼ(膵臓) |
AMY2A,EC 3.2.1.1 |
1 p21 |
澱粉の分解 |
|
アルファ2B アミラーゼ(膵臓) |
AMY2B,EC 3.2.1.1 |
1 p21 |
澱粉の分解 |
マクロアミラーゼ血症
医療においてアミラーゼ高値を呈していることは、必ずしも膵疾患(特に急性・慢性膵炎)、唾液腺疾患を意味しない。疾患を合併しない代表的なものとしてマクロアミラーゼ血症がある。これはアミラーゼと免疫グロブリンが複合体を形成し、血清アミラーゼを測定すると高値を呈するもので、臓器障害を意味しない。
同様の状態にはマクロクレアチンキナーゼ血症(マクロCK血症)がある。
脚注
1. ^ アメリカでもそのように呼ばれていた。合衆国生まれのロジャー・パルバースは小さい頃にお腹が痛くなると母親から“Take a diastase.”と言われたという。“Taka-Diastase”をそう呼んでいたのだ(『もし、日本という国がなかったら』集英社インターナショナル 2011年p.248)。
2. ^ 夏目漱石 『吾輩ハ猫デアル』 上巻、大倉書店、1905年。119頁。
3. ^ 荻原弘道 『日本栄養学史』 国民栄養協会、1960年。29頁。φ
4. ^ EC 3.2.1.1
5. ^ EC 3.2.1.2
6. ^ EC 3.2.1.3
7. ^ EC 3.2.1.68
関連項目
グルコシダーゼ
グルコシダーゼ(glucosidase)は、グリコシダーゼ(glycosidase)のうち、グルコースとのグリコシド結合を加水分解する酵素である。α-グルコシダーゼとβ-グルコシダーゼがある。
グリコシダーゼは、グルコースを含めた糖全般とのグリコシド結合を分解する酵素の総称であり、グルコシダーゼは、グルコースとのグリコシド結合を分解する酵素の個別名である。
関連項目
|
加水分解酵素 |
グリコシダーゼ |
α-グルコシダーゼ |
β-グルコシダーゼ |
β-グルコシダーゼ
β-グルコシダーゼ(β-glucosidase; EC 3.2.1.21)は糖のβ-グリコシド結合を加水分解する反応を触媒する酵素。β‐D‐グルコシドグルコヒドロラーゼ,アミグダーゼとも呼ばれる[1]。また、β-グリコシド結合を持つ代表的な糖であるセロビオースやゲンチオビオースから、しばしばセロビアーゼ、ゲンチオビアーゼとも呼ばれる。
微生物,高等植物,動物の肝臓・腎臓・小腸粘膜,カタツムリ消化液などに広く分布するが、基質特異性は起源によって異なる[1]。
α-グルコシダーゼ同様、動植物通じて広く存在し、異化代謝に関わっている。アグリコンと糖の結合も分解するが、アグリコンの構造によっては、基質が阻害剤となる場合もある。セルロースの分解に関連する酵素で、β-グルコシダーゼの活性が低いとセロビオースが蓄積し、セルロースの働きを阻害する場合がある。ただし、一般的にはセルラーゼの活性の方が低い。
β-グルコシダーゼの先天性欠損症はゴーシェ病を引き起こす[1]。
出典
1. ^ a b c β-グルコシダーゼ、『生物学辞典』、第4版、岩波書店
· IUBMB entry for 3.2.1.21(英語)
· BRENDA references for 3.2.1.21 (英語)
· PubMed references for 3.2.1.21(英語)
· PubMed Central references for 3.2.1.21(英語)
· Google Scholar references for 3.2.1.21(英語)
外部リンク
· IUBMB entry for 3.2.1.21(英語)
· KEGG entry for 3.2.1.21(英語)
· BRENDA entry for 3.2.1.21(英語)
· NiceZyme view of 3.2.1.21(英語)
· EC2PDB: PDB structures for 3.2.1.21(英語)
· PRIAM entry for 3.2.1.21(英語)
· PUMA2 entry for 3.2.1.21(英語)
· IntEnz: Integrated Enzyme entry for 3.2.1.21(英語)
· MetaCyc entry for 3.2.1.21(英語)
· Atomic-resolution structures of enzymes belonging to this class(英語)
関連項目
加水分解酵素
γ-グルタミルトランスフェラーゼ
|
γ-グルタミルトランスフェラーゼ |
|
|
識別子 |
|
|
データベース |
|
|
PDB構造 |
|
|
|
|
|
γ-グルタミルトランスフェラーゼ1 |
|
|
識別子 |
|
|
略号 |
|
|
遺伝子コード |
GGT |
|
他のデータ |
|
|
EC番号 |
|
|
γ-グルタミルトランスフェラーゼ2 |
||
|
識別子 |
||
|
略号 |
GGT2 |
|
|
遺伝子コード |
GGT |
|
|
他のデータ |
||
|
EC番号 |
||
γ-グルタミルトランスフェラーゼ(ガンマグルタミルトランスフェラーゼ、γ-glutamyltransferase; γ-GT, GGT; EC 2.3.2.2)は、グルタチオンなどのγ-グルタミルペプチドを加水分解し、他のペプチドやアミノ酸にγ-グルタミル基を転移する酵素。γ-グルタミルトランスペプチターゼ(γ-glutamyl transpeptidase; γ-GTP, GGTP)とも呼ばれる。
生体内ではそのほとんどが膜結合型酵素として存在し、膜を介したアミノ酸の移動に関与している。ヒトでは腎臓で最も活性が高く、さらに膵臓、肝臓、脾臓、小腸、精巣、前立腺など広く全身に分布する。
肝臓では、肝細胞のミクロソーム分画で産生され、細胆管、毛細胆管などの細胞膜に移動して機能している。これが閉塞性黄疸、肝癌、アルコール性肝障害など肝・胆道系の疾患で誘導され、逸脱酵素として血中に流出する。このため、血中のγ-グルタミルトランスフェラーゼ活性は肝機能の指標として利用されている。
関連項目
外部リンク
gamma-Glutamyltransferase - MeSH、米国国立医学図書館、生命科学用語シソーラス (英語サイト)
アスパラギン酸アミノ基転移酵素
|
アスパラギン酸トランスアミナーゼ |
|
|
Escherichia coliのアスパラギン酸トランスアミナーゼのリボンモデル。中央の分子は補因子のピリドキサールリン酸[1] |
|
|
識別子 |
|
|
データベース |
|
|
PDB構造 |
|
|
|
|

アスパラギン酸アミノ基転移酵素(アスパラギンさんアミノきてんいこうそ、Aspartate Aminotransferase, ART ; EC 2.6.1.1)は、アスパラギン酸とα-ケトグルタル酸をオキサロ酢酸とグルタミン酸に相互変換する酵素である。AST (Aspartate transaminase) またはGOT(Glutamic Oxaloacetic Transaminase:グルタミン酸オキサロ酢酸トランスアミナーゼ)とも呼ばれる。
主にミトコンドリア内で働く m-AST と細胞質基質で働く s-AST に分類される。
人体では、肝細胞をはじめとして赤血球、心筋、骨格筋などに分布する。これらの細胞が破壊された場合には血液中に流出するため、血中濃度を測定することで肝障害などの程度を知ることができる(詳細は逸脱酵素を参照)。
臨床検査におけるAST
逸脱酵素としての性質から、肝機能障害の程度を評価する目的で血清中のAST濃度測定が行われる。ただし、肝障害のマーカーとしては、肝細胞が破壊し尽くされるとむしろ流出量は低下する点と、肝臓以外の障害(心筋梗塞や溶血性貧血)でも上昇しうる点に留意すべきである。肝臓に特異的という点では、ALT (GPT) も同時に評価することが有用となる。
基準値
単位は IU/l(国際単位/l)で示され、10 - 40程度が基準値となる。 但し、基準値内であれば「正常である」という事は出来無い。
異常値
肝炎、脂肪肝、肝硬変、肝腫瘍、などの肝疾患ではAST、ALTの上昇が特徴的であり、100以上、ときに500以上を示す。なかでも、アルコール性肝炎や肝硬変、肝腫瘍ではASTの上昇が目立ち、ウイルス性肝炎や脂肪肝ではALTの上昇が目立つとされている。膠原病の1種である多発性筋炎 (PM)、皮膚筋炎 (DM) でもAST (GOT) の上昇を認める。
このほか、AST上昇時には心筋梗塞、溶血性貧血などが鑑別疾患にあがる。採血時の溶血の可能性も考慮する必要がある。
出典
1. ^ PDB 1AAMAlmo SC, Smith DL, Danishefsky AT, Ringe D (March 1994). “The structural basis for the altered substrate specificity of the R292D active site mutant of aspartate aminotransferase from E. coli”. Protein Eng. 7 (3): 405–12. doi:10.1093/protein/7.3.405. PMID 7909946.
関連項目
クレアチンキナーゼ
|
creatine kinase MM, dimer, Human. |

クレアチンキナーゼ(Creatine Kinase、CK)、CPK(クレアチンホスホキナーゼ、Creatine PhosphoKinase)は、動物が持つ酵素で、筋肉の収縮の際にエネルギー代謝に関与している。EC番号2.7.3.2。
働きは、クレアチンとATPからクレアチンリン酸とADPが生成する反応の媒介である。骨格筋や心筋など、興奮性を持つ細胞に分布している。
臨床検査
CKは骨格筋・心筋が障害を受けた際に血液中へ流出する逸脱酵素として臨床上重要である。 心筋梗塞、筋炎、筋ジストロフィーなど心筋障害・筋疾患で血中濃度が上昇する。ただし、激しい運動などでも筋線維が壊れるためCKの上昇がみられることがある。
単位はIU(国際単位)/L。正常値は男性の方が高く(筋肉量の違いによる)、男性で30〜190 IU/L、女性で20〜150 IU/L程度とされている。
アイソザイム
CKは二つのサブユニットからなる二量体の蛋白質である。このサブユニットには2種類あり(B:脳型、M:筋型)、この組み合わせによって3種類のCK(MM、BB、MB)がアイソザイムとして存在する。骨格筋にはMM型、心筋にはMB型が多いため、原因不明のCK上昇ではこのアイソザイム比率を分析することで有意義な所見を得られることがある。また、急性心筋梗塞が疑われる際にはCK-MBの測定が診断の裏付け、および重症度の指標となる。
心筋梗塞の発症後、CK値が上昇するには若干のタイムラグがある(数時間後から上昇)。急性期の対応では、確定診断のためにはCK-MBよりも早く上昇し、やはり特異性の高いトロポニンT値が重視される。
CK-MBでも骨格筋由来が正常でも5%程度認められることが知られている。そのためCK上昇時には比率を調べる必要がある。また骨格筋からも筋再生時にはCK-MBが産出されることが知られている。この場合はトロボニンTなども同様に産出される。特に皮膚筋炎、多発性筋炎の活動期にはCKの25%がCK-MBとなることもある。上記疾患の合併症に心筋炎が認められることもあるため心臓超音波検査の併用が必要である。
マクロクレアチンキナーゼ血症
免疫グロブリンとクレアチンキナーゼが結合し、検査上高値となる症候があり、マクロクレアチンキナーゼ血症(マクロCK血症)と呼ばれる。ほとんどは疾患を意味するものではないが、時に悪性腫瘍や膠原病によるマクロCK血症もあり、注意を要する。
|
||||||||||||||||||
<img src="//ja.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1x1" alt="" title="" width="1" height="1" style="border: none; position: absolute;" />
「https://ja.wikipedia.org/w/index.php?title=クレアチンキナーゼ&oldid=61838504」から取得
カテゴリ:
け
形成性操作タンパク質
形成性操作タンパク質(けいせいせいそうさタンパクしつ、ディリジェントタンパク質、英: dirigent protein)は、他酵素によって合成される化合物の立体化学を決定づけるタンパク質である[1]。最初の形成性操作タンパク質はForsythia × intermediaにおいて発見された。このタンパク質はコニフェニルアルコール単量体からの(+)-ピノレシノールの立体選択的生合成を指示することが明らかにされている[2]。
|
|
-Pinoresinol_Biosynthesis.png)
リグナンの生合成は酸化酵素 (oxidative enzyme) によって触媒される[3]。試験管内では、反応は二量体化合物の不均一混合物を与える[4]。反応の間に形成性操作タンパク質が存在すると、1種類の化合物の1種類の立体異性体が高い選択性で得られる。形成性操作タンパク質それ自身は酸化的ラジカル形成活性を持たないように見える、酸化酵素がなければ反応は起こらない[5]。
近年、2番目のエナンチオ相補的形成性操作タンパク質がシロイヌナズナ(Arabidopsis thaliana)から発見された[6]。この酵素は (−)-ピノレシノールのエナンチオ選択的合成を指示する。
活性
リグナンの生合成において、酸化酵素は水素原子プロトン共役型電子移動によってモノリグノールから水素原子を取り除きラジカル中間体を形成する。これらの中間体は次にラジカル停止反応に連結し、リグナンとして知られる様々な二量体の中の一つを形成する[7]。酸化酵素存在下でのコニフェリルアルコール(一般的なモノリグノール)のin vitro反応では、様々な濃度の様々な異なる二量体が得られる[8]。Forsythia × intermedia由来の形成性操作タンパク質が存在すると、(+)-ピノレシノールの生成が著しく強化され、その他の生成物は極めて少なくなる。この強化が非常に明白なため、形成性操作タンパク質は (+)-ピノレシノールのみを生産し、様々な不均一混合物が生成するタンパク質が介在しない連結反応と競合している、との仮説が立てられている[9]。これは、異なる濃度の形成性操作タンパク質存在下で生成する様々な混合物を分析することによって確かめられている。この立体選択性が達成される機構は現時点でよく分かっていない。しかしながら、酸化酵素がなければ反応が進行しないため、形成性操作タンパク質自身はラジカルを形成するためのコニフェリルアルコールの酸化を触媒しないように見える。
|
|

Forsythia intermedia由来の形成性操作タンパク質存在下で、(+)-ピノレシノールの生成が大幅に強化され、その他の二量体生成物の生成は阻害される。
Forsythia intermedia由来の形成性操作タンパク質の活性はコニフェリルアルコール特異的である[10]。p-クマリルアルコールやシナピルアルコールといったその他のモノリグノールが形成性操作タンパク質存在下で酸化酵素とin vitroで反応した場合、形成性操作タンパク質非存在下での実験と区別できない生成物の不均一な混合物を与える。
構造
円偏光二色性実験により、Forsythia intermedia由来の形成性操作タンパク質の二次構造は主にβシートとループ構造から成ることが明らかにされている。三次構造は解かれていないが、タンパク質は二量体であることが確かめられている。それぞれの単量体は単一のコニフェリルアルコール結合部位を有し、全部で2つの結合部位が存在する[11]。1分子のコニフェリルアルコールがそれぞれの部位に結合できるため、2分子間の反応構造が限定されることによって、(+)-ピノレシノールの生成が増加し、その他の生成物の生成が阻害される。
脚注
1. ^ Davin LB, Wang HB, Crowell AL, et al. (1997). “Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center”. Science 275 (5298): 362–6. doi:10.1126/science.275.5298.362. PMID 8994027.
2. ^ Davin LB, Wang HB, Crowell AL, et al. (1997). “Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center”. Science 275 (5298): 362–6. doi:10.1126/science.275.5298.362. PMID 8994027.
3. ^ Ward R.S. (1982). “The synthesis of lignans and neolignans”. Chemical Society Reviews 11: 75–125. doi:10.1039/CS9821100075.
4. ^ Fournand D, Cathala B, Lapierre C (January 2003). “Initial steps of the peroxidase-catalyzed polymerization of coniferyl alcohol and/or sinapyl aldehyde: capillary zone electrophoresis study of pH effect”. Phytochemistry 62 (2): 139–46. doi:10.1016/S0031-9422(02)00573-3. PMID 12482448.
5. ^ Davin L.B., Lewis N.G. (August 2005). “Dirigent phenoxy radical coupling: advances and challenges”. Current Opinion in Biotechnology 16 (4): 398–406. doi:10.1016/j.copbio.2005.06.010.
6. ^ Pickel B, Constantin M-A, Pfannsteil J, Conrad J, Beifuss U, Schaffer A (March 2007). “An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols”. Angewandte Chemistry 53 (4): 273–284. doi:10.1007/s10086-007-0892-x.
7. ^ Sarkanen, Simo; Lewis, Norman (1998). Lignin and lignan biosynthesis. Columbus, OH: American Chemical Society. ISBN 0-8412-3566-X.
8. ^ Fournand D, Cathala B, Lapierre C (January 2003). “Initial steps of the peroxidase-catalyzed polymerization of coniferyl alcohol and/or sinapyl aldehyde: capillary zone electrophoresis study of pH effect”. Phytochemistry 62 (2): 139–46. doi:10.1016/S0031-9422(02)00573-3. PMID 12482448.
9. ^ Halls SC, Davin LB, Kramer DM, Lewis NG (2004). “Kinetic study of coniferyl alcohol radical binding to the (+)-pinoresinol forming dirigent protein”. Biochemistry 43 (9): 2587–95. doi:10.1021/bi035959o. PMID 14992596.
10. ^ Kim, M.K., Jeon J-H, Fujita M, Davin L.B., Lewis N.G. (2002). “The western red cedar (Thuja plicata) 8-8′ DIRIGENT family displays diverse expression patterns and conserved monolignol coupling specificity”. Plant Molecular Biology 49 (2): 199–214. doi:10.1023/A:1014940930703.
11. ^ Halls SC, Lewis NG (July 2002). “Secondary and quaternary structures of the (+)-pinoresinol-forming dirigent protein”. Biochemistry 41 (30): 9455–61. doi:10.1021/bi0259709. PMID 12135368.
β-グルコシダーゼ
β-グルコシダーゼ(β-glucosidase; EC 3.2.1.21)は糖のβ-グリコシド結合を加水分解する反応を触媒する酵素。β‐D‐グルコシドグルコヒドロラーゼ,アミグダーゼとも呼ばれる[1]。また、β-グリコシド結合を持つ代表的な糖であるセロビオースやゲンチオビオースから、しばしばセロビアーゼ、ゲンチオビアーゼとも呼ばれる。
微生物,高等植物,動物の肝臓・腎臓・小腸粘膜,カタツムリ消化液などに広く分布するが、基質特異性は起源によって異なる[1]。
α-グルコシダーゼ同様、動植物通じて広く存在し、異化代謝に関わっている。アグリコンと糖の結合も分解するが、アグリコンの構造によっては、基質が阻害剤となる場合もある。セルロースの分解に関連する酵素で、β-グルコシダーゼの活性が低いとセロビオースが蓄積し、セルロースの働きを阻害する場合がある。ただし、一般的にはセルラーゼの活性の方が低い。
β-グルコシダーゼの先天性欠損症はゴーシェ病を引き起こす[1]。
出典
1. ^ a b c β-グルコシダーゼ、『生物学辞典』、第4版、岩波書店
· IUBMB entry for 3.2.1.21(英語)
· BRENDA references for 3.2.1.21 (英語)
· PubMed references for 3.2.1.21(英語)
· PubMed Central references for 3.2.1.21(英語)
· Google Scholar references for 3.2.1.21(英語)
外部リンク
· IUBMB entry for 3.2.1.21(英語)
· KEGG entry for 3.2.1.21(英語)
· BRENDA entry for 3.2.1.21(英語)
· NiceZyme view of 3.2.1.21(英語)
· EC2PDB: PDB structures for 3.2.1.21(英語)
· PRIAM entry for 3.2.1.21(英語)
· PUMA2 entry for 3.2.1.21(英語)
· IntEnz: Integrated Enzyme entry for 3.2.1.21(英語)
· MetaCyc entry for 3.2.1.21(英語)
· Atomic-resolution structures of enzymes belonging to this class(英語)
関連項目
· 加水分解酵素
こ
合成酵素
合成酵素(ごうせいこうそ)とは
日本語表記では次の2つのグループに属する酵素の一部が合成酵素と呼ばれる。合成酵素といった場合は特定の酵素グループを意味しない点を留意すべきである。
リアーゼ〈EC番号4群〉に属する一部酵素。ATP加水分解を必要としない。
リガーゼ〈EC番号6群〉に属する酵素。ATP加水分解を必要とする。
酵素栄養学
酵素栄養学(こうそえいようがく、英: enzyme nutrition)とは、酵素が重要な栄養素だとみなす理論である。エドワード・ハウエルが1946年に専門書[1] を、1980年と1985年に一般向けの著書を出版して知られるようになった。生の食品の摂取を推奨しており、ローフーディズムの主要な根拠のひとつとなっている。現代の生化学に反するという批判もある。
エドワード・ハウエルの主張
エドワード・ハウエルは、「潜在酵素」、「食物酵素」という言葉でこの理論を説明した[2]。
「潜在酵素」とは、体内で消化のほか、様々な生体の活動に用いられる酵素を総合的にとらえた概念である。この潜在酵素は、生物の一生で使われる総量に上限があり、これが消耗されすぎると病気の原因となり、寿命は縮むと考えられる。一方で食品に含まれる酵素を「食物酵素」と呼んだ。食物酵素の多い食事をすると、食物酵素が食品の消化を助け、人体自身の消化酵素の分泌が少なくてすむために、潜在酵素の消費を抑えることができると考えられた。さらに、酵素には「生命エネルギー」が含まれているとし、酵素の多い食物を取ることは病気を予防し、寿命を延ばすエネルギーの補充の効果があるとした。
自然の状態の食品には、元となった生物由来の酵素が含まれている。多くの動物は複数の胃を持っているが、反芻胃などで食物や微生物の酵素を利用して「事前消化」を行い、最後の胃で自身が作り出した酵素を利用する。ハウエルは人間でも胃のはじめの方の部分はこうした役割を持っていると主張した。
しかし、酵素の多くは加熱によって失活する。こうして酵素の活性を失った食品を食べた場合、はじめの胃で食物酵素による事前消化が行われず、身体で作り出した酵素を使用することになり、消化酵素を作り出している膵臓などにより負担をかけることになる。これが病気の原因となるので、食物の多くを生で食べることをすすめた。さらに、今の時代は食品の調理や加工によって酵素が活性を失った食品も多いので、酵素のサプリメントを摂取することも推奨した。この考え方はペットフードにも応用され、ペット用のサプリメントも市販されている。
また、酵素を多く含んでるので、発酵食品を薦めた。例えば、味噌では麹菌が作るアミラーゼやリパーゼをはじめとした各種の酵素が蓄積されている[3]。
酵素を摂り込むために、食品は生で食べることを勧めているが、穀物や豆などの種子は例外としている。酵素の働きを抑制する「酵素抑制物質」を含み、そのまま食べると害になるためだ。そこで、種子は発芽させて「スプラウト」の状態にする。発芽する過程で、酵素抑制物質は消滅し、しかも酵素活性が高まり、優れた酵素食材となる。
日本では、鶴見隆史、新谷弘実らが類似した主張をしている。鶴見はスプラウトに絶大な健康効果があるとしている。新谷はすべての酵素の元となる物質が体内に蓄積されていると想定し、「ミラクル・エンザイム」と名づけ、その利用を節約することで長生きができると主張し、シンヤビオジマを提唱している[4]。新谷はまた、酵素を構成するアミノ酸には、「記憶」があり、食物中の酵素が分解され、消化吸収された後にも、体内で酵素に再構成されると主張している。そのため、酵素を多く含んだ食品を摂取することは消化の助けになるだけでなく、体内で代謝に使われる酵素を補充する意義もあるとしている。
アメリカでは、テキサス州ヒューストンにてディッキー・フュラー博士が酵素を用いた診療を行っている[5]。
一般的な生理学・分子生物学等との矛盾点
1985年のハウエルの著書では、引用された科学文献のほとんどが20世紀前半の研究であるといった問題点があり、時代遅れであると指摘されている[6]。
「潜在酵素」に該当する事実も発見されていない。一般的な分子生物学や生化学の知見では、多種類の酵素の遺伝子は、それぞれ個別に制御されているとされており、総合的な酵素生産に上限があるという事実は発見されていない。また、酵素は触媒であるため、化学反応後にも消耗されることはない[7]。
「食物酵素」が食品の「事前消化」に重要であるという考え方も一般には考えづらい。生物体にはその生物の生存のために多種類の酵素が含まれているが、それが食材となったときに消化に大きく寄与する、という考え方は一般的ではない。酵素が活性を発揮するためには、pHや温度、反応溶液の塩濃度等の条件が厳密に定められており、強酸性の胃の中では、食物自身の持つ酵素は大部分が速やかに変性してしまう[6]。草食動物が「事前消化」を行う際には、消化管に共生する微生物の働きが重要であるが、餌中の酵素の影響が多大であるという報告はない[8]。草食動物と異なり、人間の胃にそのような微生物は共生しておらず、「事前消化を行う前半部分」があるということも解剖学的に報告されてはいない。
アメリカでは2003年にFDAは、消化酵素のサプリメントの販売者に対し、科学的根拠がないとして警告を行った[9]。
脚注
1. ^ Edward Howell (1946). The status of food enzymes in digestion and metabolism. Chicago: National enzyme company. OCLC 5639180.
2. ^ 小池里予、小池英 『ホリスティック健康学・ホリスティック栄養学入門』 ホリスティック栄養学研究所、2004年、268頁。ISBN 978-4990196400。
3. ^ 今井誠一 『味噌 : 色・味にブレを出さない技術と販売』 農山漁村文化協会〈食品加工シリーズ〉、2002年、26-27頁。ISBN 4-540-01151-0。
4. ^ 新谷弘実 『病気にならない生き方 : ミラクル・エンザイムが寿命を決める』 サンマーク出版、2005年。ISBN 4-7631-9619-7。
5. ^ ディッキー・フュラー 『病気を癒し、老化を防ぐ 酵素の治癒力』 竹内 進一郎訳、現代書林、2011年。ISBN 978-4774513393。
6. ^ a b “Do 'Food Enzymes' Enhance Digestive Efficiency, Longevity?”. Beyond Vegetarianism. 2013年10月2日閲覧。
7. ^ Donald Voet、Judith G.Voet 『ヴォート生化学 上』 田宮信雄ほか訳、東京化学同人、2005年(原著2002年)、第3版。ISBN 4-8079-0607-0。[要ページ番号]
8. ^ 『反芻動物の栄養生理学』 小原嘉昭編、佐々木康之監修、農山漁村文化協会、1998年。ISBN 4-540-98049-1。
9. ^ Stephen Barrett (2003年3月11日). “"Enzyme Deficiency"”. Quackwatch. 2013年10月2日閲覧。
参考文献
- エドワード・ハウエル 『キラー・フード : あなたの寿命は「酵素」で決まる』 川喜田昭雄監訳・瀬野川知子訳、現代書林(原著1985年)。ISBN 4-7745-0097-6。(原著 ENZYME NUTRITION, 1985)
- エドワード・ハウエル 『食物酵素のbaka力』 今村光一訳、ヘルス・ビジネス・マガジン社、2002年(原著1994年)。(原著 FOOD ENZYMES FOR HEALTH & LONGEVITY, 1994)
- エドワード・ハウエル 『医者も知らない酵素の力』 今村光一訳、中央アート出版社、2009年(原著1994年)、改版。ISBN 978-4-8136-0535-5。(原著 FOOD ENZYMES FOR HEALTH & LONGEVITY, 1994)
関連項目
外部リンク
<img src="//ja.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1x1" alt="" title="" width="1" height="1" style="border: none; position: absolute;" />
「https://ja.wikipedia.org/w/index.php?title=酵素栄養学&oldid=67777795」から取得
カテゴリ:
酵素前駆体
酵素前駆体(こうそぜんくたい zymogen チモーゲン)とは、不活性な酵素前駆体のことである。
酵素前駆体が活性を持つ酵素に変化するには、加水分解や構造変化などの生化学的変化によって活性部位が働ける状態になる必要がある。 なかでも、酵素前駆体の一部がプロテアーゼによって切断される例は多く、活性化の過程で遊離したペプチド鎖は活性化ペプチドと呼ばれる。
酵素前駆体の例[編集]
酵素前駆体の例として、以下のようなものがある。
トリプシノーゲン: 消化酵素 トリプシン の前駆体
キモトリプシノーゲン
消化酵素 キモトリプシン の前駆体
ペプシノーゲン
消化酵素 ペプシン の前駆体
プロエラスターゼ
消化酵素 エラスターゼ の前駆体
プロリパーゼ
消化酵素 リパーゼ の前駆体
血液凝固系の酵素の大半
カスケード反応を形成している
プラスミノゲン
線溶系の酵素 プラスミン の前駆体
補体系の酵素の一部
カスパーゼ
アポトーシスを実行するシグナルカスケードを構成している
このうち、トリプシン、キモトリプシン、エラスターゼ、凝固系酵素、プラスミン、補体系酵素はセリンプロテアーゼである。カスパーゼはシステインプロテアーゼであり、ペプシンはアスパラギン酸プロテアーゼである。
なお、活性化された酵素を、別の酵素が修飾して不活性化する過程も存在する。例えば、凝固系のプロテインCは、活性型第V因子や活性型第VIII因子を分解して不活性化する。
意義[編集]
活性型の酵素をそのまま合成するのではなく、まず不活性型の前駆体として合成しておき、後で活性化するのは、次のような意義があると考えられている。
まず、迅速な調節が可能となることである。遺伝子の転写、mRNAの翻訳 には数十分〜数時間がかかるため、出血のような緊急事態に対応するには間に合わない。活性を持たない前駆体をあらかじめ用意しておけば、必要が生じたときにすぐに活性化して使うことができる。
次に、反応を増幅できることである。凝固系・補体系・アポトーシスなどの経路は、活性化された酵素が次の段階の酵素を活性化するというカスケード反応になっている。一つの酵素は無数の基質を反応させることができるので、小さな入力から大きな出力(反応)を得ることができる。
また、酵素活性を、時間的・空間的に限定するという意味もある。つまり、必要なときに、必要な場所でのみ、酵素を活性化させるのである。例えば、消化酵素は食物を分解して吸収できるようにする上で不可欠なものだが、自己の組織をも分解しかねない危険な存在である。そこで、酵素を前駆体として合成して不活性な状態で貯蔵しておき、食事後に消化管内に分泌されて初めて活性化するようにしている。急性膵炎は、この仕組みが破綻した状態といえる。膵臓の中で消化酵素が活性化され、膵臓と周辺臓器が溶かされてしまう重篤な疾患である。播種性血管内凝固症候群 も、凝固系と線溶系の酵素活性のバランスが崩れた状態といえる。
酵素阻害剤
酵素阻害剤(こうそそがいざい)とは、酵素分子に結合してその活性を低下または消失させる物質のことである。酵素阻害剤は一般に生理活性物質であり、毒性を示すものもあるが、病原体を殺したり、体内の代謝やシグナル伝達などを正常化したりするために医薬品として利用されるものも多い。また殺虫剤や農薬などに利用される種類もある。
酵素に結合する物質すべてが酵素阻害剤というわけではなく、逆に活性を上昇させるもの(酵素活性化剤)もある。
酵素阻害剤の作用には、酵素の基質が活性中心に入って反応が始まるのを阻止するもの、あるいは酵素による反応の触媒作用を阻害するものがある。また酵素に可逆的に結合するもの(濃度が下がれは解離する)と、酵素分子の特定部分と共有結合を形成して不可逆的に結合するものとに分けられる。さらに阻害剤が酵素分子単独、酵素・基質複合体、またその両方に結合するかなどによっても分類される。
生体内にある物質が酵素阻害物質になることもある。例えば、代謝経路の途中にある酵素では、下流の代謝産物により阻害されるものがあり(フィードバック阻害)、これは代謝を調節する機構として働いている。さらに、生物体内にあって生理的機能を持つ酵素阻害タンパク質もある。これらはプロテアーゼやヌクレアーゼなど、生物自身に害を及ぼしうる酵素を厳密に制御する機能を持つものが多い。
酵素阻害剤には、基質と同様に酵素に対する特異性がある場合が多い。一般に医薬品としての阻害剤では、特異性の高い方が毒性・副作用が少ないとされる。また抗菌薬や殺虫剤に求められる選択毒性を出すためにも高い特異性が必要である。
酵素阻害剤の種類]
酵素阻害剤には可逆的および不可逆的なものがある。可逆的阻害剤は作用機序により次のように分類される。
基質阻害
酵素・基質複合体に結合して反応の進行を妨げる。
競争(拮抗)阻害
基質と同じ部位に競合的に結合して反応開始を妨げる。
非競争(非拮抗)阻害
酵素または酵素・基質複合体の、基質と別の部位に結合して反応の進行を妨げる。
不競争(不拮抗)阻害
酵素・基質複合体のみの、基質と別の部位に結合して反応の進行を妨げる。
混合型阻害
これらの分類は反応速度パラメーターを測定することで明らかにできる。詳細については酵素反応速度論およびミカエリス・メンテン式の項を参照されたい。
その他の阻害
酵素以外の一般のタンパク質に対しても、同様にその機能を阻止する阻害剤がある。これらも医薬品などに応用される。
ホルモンや神経伝達物質など内在性の生理活性物質(リガンド)に対しては、それを特異的に結合するタンパク質である受容体が存在する。受容体に結合することによりリガンドの機能を阻止する拮抗的阻害剤は、特にアンタゴニストまたは遮断薬と呼ばれる。
利用例
医薬品などとして利用される酵素または一般タンパク質の阻害剤には、次に挙げるようなものがある。
β-ラクタム系抗生物質
サルファ薬
ノイラミニダーゼ阻害薬
非ステロイド系抗炎症薬
分子標的治療薬
スタチン
ホスホジエステラーゼ阻害薬
ACE阻害薬
モノアミン酸化酵素阻害薬
コリンエステラーゼ阻害薬
プロトンポンプ阻害薬
選択的セロトニン再取り込み阻害薬
ブドウ糖吸収阻害薬(αグルコシダーゼ阻害薬)
DPP-4阻害薬
このほか、アンタゴニストで医薬品として利用されるものも数多い。
関連項目[編集]
酵素
酵素反応
酵素反応速度論
ミカエリス・メンテン式
アロステリック効果
ケミカルバイオロジー
表 話 編 歴
タンパク質:酵素
トピックス
活性部位 アロステリック効果 結合部位 触媒三残基 補酵素 補因子 共同性 EC番号 酵素反応 酵素阻害剤 酵素反応速度論 ミカエリス・メンテン式
タイプ
EC1 酸化還元酵素 EC2 転移酵素 EC3 加水分解酵素 EC4 リアーゼ EC5 異性化酵素 EC6 リガーゼ
酵素反応
出典: フリー百科事典『ウィキペディア(Wikipedia)』
酵素反応(こうそはんのう)とは、酵素が触媒する生化学反応である。
目次
[非表示]
酵素反応速度[編集]
詳細は「酵素反応速度論」を参照
日本工業規格に「酵素は選択的な触媒作用を持つタンパク質を主成分とする生体高分子物質」 (JIS K 3600-1310) と定義されているように触媒として利用されるが、化学工業などで用いられる典型的な金属触媒とは反応の特性が異なる。
第一に酵素反応の場合、基質濃度 [S] が高くなると反応速度が飽和する現象が見られる。酵素の場合、基質濃度を高く変えると、反応速度は飽和最大速度 Vmax へと至る双曲線を描く。一方、金属触媒の場合、反応初速度 [ν] は触媒濃度に依存せず基質濃度 [S] の一次式で決定される。
このことは、酵素と金属触媒との粒子状態の違いによって説明できる。金属触媒の場合、触媒粒子の表面は金属原子で覆われており、無数の触媒部位が存在する。それに対して酵素の場合、酵素分子が基質に比べて巨大な場合が多く、活性中心を高々1か所程度しか持たない。そのため金属触媒に比べて、基質と触媒(酵素)とが衝突頻度しても(活性中心に適合し)反応を起こす頻度が小さい。そして基質濃度が高まると、少ない酵素の活性中心を基質が取り合うようになるので、飽和現象が生じる。このように酵素反応では、酵素と基質が組み合った基質複合体を作る過程が反応速度を決める律速過程になっていると考えられる。
また、酵素反応において基質複合体の濃度 [ES] の時間応答を調べると、系に酵素を投入しても、基質複合体を形成するのに時間がかかるので、濃度 [ES] は緩やかに上昇する。その後は生成物 P の産生が始まって濃度 [ES] は定常状態となる。このように基質複合体濃度の立ち上がりの前の定常状態とその後の定常状態が観測されるのも、酵素反応の特徴である[1]。
酵素反応の定式化[編集]
1913年L・ミカエリスとM・メンテンは酵素によるショ糖の加水分解反応を測定し、「鍵と鍵穴」モデルと実験結果から酵素基質複合体モデルを導き出し、酵素反応を定式化した。このモデルによると、酵素を用いた系では以下の式で反応が進行する。
酵素 (E) + 基質 (S) 酵素基質複合体 (ES) → 酵素 (E) + 生産物 (P)
 酵素基質複合体
酵素基質複合体すなわち、酵素反応は、酵素と基質が一時的に結びついて酵素基質複合体を形成する第1の過程と、酵素基質複合体が酵素と生産物とに分離する第2の過程とに分けられる。
この理論から導かれるミカエリス・メンテン式によって、酵素反応の反応速度が求められる。ミカエリスとメンテンによる最初の理論は E + S と ES との間の化学平衡を仮定しており、ゆっくりと生成物へと反応が進行する場合の近似だったが、のちにブリッグスとホールデンがより一般的な定常条件を仮定し、その場合でも同様の式が成り立つことを示した。
酵素と基質が酵素基質複合体を形成する過程(上記の式の第1の過程)は、可逆過程として扱うことができる。この反応が定常状態である時の平衡定数はミカエリス・メンテン定数と呼ばれる酵素反応の重要なパラメータで、 Km と表記される。この定数は酵素と基質の親和性を表すパラメータであり、以下の性質を持つ:
- Km値が低いと酵素と基質の親和性は高く、素早く複合体形成するが生成反応の進行は遅い。
- Km値が高いと酵素と基質の親和性は低く、ゆっくりと複合体形成するが生成反応の進行は素早い。
なお、Km の実測値は、酵素反応の反応速度が最大速度Vmaxの2分の1となるときの基質濃度と同じ値になる。
また、Vmax と関連した分子活性 kcat という値が存在する。これはタンパク質1分子あたり、1秒間に何個の基質を触媒するか、と言うパラメータである。式は以下のように表される。
kcat = 基質分子濃度 (M)/酵素分子濃度 (M) × 秒
ここで右辺は分子と分母に濃度の単位を持つのでこれを約すと、kcat は s−1 という単位で現される。例を挙げれば、酵素1分子あたり1秒間に100個の基質分子を触媒すれば 100 s−1 となる。炭酸脱水酵素には極めて活性の高いものがあるが、この酵素は1秒当たり百万個の二酸化炭素を炭酸イオンに変化させる (kcat = 106 s−1)。
阻害様式と酵素反応速度[編集]
酵素の反応速度は、基質と構造の似た分子の存在や、後述のアロステリック効果により影響を受ける(阻害される)。阻害作用の種類によって、酵素の反応速度の応答の様式(阻害様式)が変わる。そこで、反応速度や反応速度パラメーターを解析して阻害様式を調べることで、逆にどのような阻害作用を受けているかを識別することができる。
阻害様式は大きく分けると次のように分類される:
酵素の反応速度曲線を阻害剤のない原系の場合を青線、阻害剤の存在する系を赤線で示すと次のようになる:
拮抗阻害の場合は Vmax は移動せず、Km が移動する。一方、非拮抗阻害の場合は Km は移動せず Vmax が移動する。混合型阻害の場合は図に示さないが両方の寄与が見られる。
多くの場合、阻害剤が基質に類似している場合は拮抗阻害を示す。またアロステリック阻害は拮抗的ではない阻害に該当する。それぞれの阻害様式の場合の定式化は記事ミカエリス・メンテン式に詳しい。
酵素反応の活性化エネルギー[編集]
一般に化学反応の進行する方向は基質や生成物の濃度、温度・圧力など(熱力学的状態)によって決定付けられる。言い換えると、化学反応は化学ポテンシャルが小さくなる方向に進行し、反応速度は反応の活性化エネルギーが高いか否かに大きく左右される(記事 化学平衡や反応速度論に詳しい)。
酵素反応は触媒反応で、化学反応の一種なので、その性質は同様である。ただし、一般に触媒反応は化学反応の中でも活性化エネルギーが低いのが通常であるが、酵素反応の活性化エネルギーは特に低いものが多い。
|
触媒の活性化エネルギー比較[2] |
||
|
反応名 |
触媒/酵素† |
|
|
H2O2の分解 |
(なし) |
18,000 |
|
白金コロイド |
11,000 |
|
|
5,000 |
||
|
ショ糖の加水分解 |
H+ |
26,500 |
|
サッカラーゼ†(酵母) |
11,500 |
|
|
カゼインの加水分解 |
HCl aq. |
20,000 |
|
キモトリプシン†(Trypsin) |
12,000 |
|
|
酢酸エチルの加水分解 |
H+ |
13,200 |
|
4,200 |
||
一般に活性エネルギーが15,000cal/molから10,000cal/molに低下すると、反応速度定数はおよそ4.5×107倍になる。
反応機構モデル[編集]
酵素の基質特異性はなぜ発揮されるのか、活性化エネルギーをいかにして下げるのかなど、無機触媒や酸塩基触媒などと違う基本的特性を生み出す酵素反応の機構については、未だ統一的な解答が得られたとはいえない。しかし今日では、構造生物学の発展や組み換えタンパク質作成による変異導入などのテクニックを用いることにより、その片鱗が明らかにされつつある。
基質の結合[編集]
酵素には触媒反応に中心的な役割を果たす活性中心という部分が存在し、その近傍に、基質を結合して触媒のために安定化させる基質結合部位を持っている。酵素の活性中心や基質結合部位はタンパク質の三次構造の裂け目(クラフト)の内部に存在することが多く、そこにアミノ酸の側鎖が適当に配置されている。乳酸デヒドロゲナーゼではN末端から数えて250番目のリシンが NAD+ の結合に関係している。また、セリンプロテアーゼと呼ばれるタンパク質分解酵素の一群は基質の結合部位にセリンが存在する。
基質結合部位に基質が結合するにあたっては、基質は誘導的に酵素に結合すると考えられている。つまり、酵素と基質が結合した状態が基質にとってはエントロピー的に減少するということである。この考え方をエントロピー・トラップという。このエントロピー・トラップにより基質は誘導的に基質結合部位に結合していく。一般に基質濃度を上昇させることも化学反応を起こすための1つの条件であるが、酵素は低濃度基質条件下でも効率よく触媒する。その理由は、エントロピー・トラップによって活性中心付近における基質の濃度が上昇するためだと考えられている。
酵素の内の酸塩基触媒部位[編集]
化学反応を起こさせるには pH が極端な状態に基質を置くのが有効な場合があり、酵素内でも同様の変化が起きていると考えられている。特に加水分解や脱水素反応は酸塩基触媒部位によるプロセスが重要であり、キモトリプシンなどでは以下のプロトンの伝達のモデルが考えられている。
- His57 がプロトンを負に荷電した Asp102 に譲渡する
- His57 が塩基となり、活性中心の Ser195 からプロトンを奪う
- Ser195 が活性化されて(負に荷電して)基質を攻撃する
- His57 がプロトンを基質に譲渡する
- Asp102 から His57 がプロトンを奪い 1. の状態に戻る
ここで His などはタンパク質を構成するアミノ酸残基の3文字略号を示し、右肩の数字は N 末端からの番号を表す。酵素の中で、酸塩基触媒として最も作用するのはヒスチジンである。ヒスチジンは等電点が pH 6 であり、生理的な条件に極めて近い。ヒスチジンはプロテアーゼ以外にも脱水素酵素の活性中心を担当している場合が多い。
遷移状態と抗体酵素[編集]
酵素反応において、酵素基質複合体から生成物へと変化する過程では、原子間の結合距離や角度などが変形した分子構造となる遷移状態や反応中間体を経由する。
分子の内部ポテンシャルエネルギーは原子間の結合距離や角度に応じて様々に変化するが、通常見られる分子においてはポテンシャルエネルギーが極小となる結合距離や角度をとっている。酵素反応の基質もポテンシャルエネルギーは極小となっている。そして、酵素反応が進行する過程(遷移状態)では、原子間の結合距離や角度の変化を伴い、その変化によりポテンシャルエネルギーは一時的に増大するが、反応が進むと再び減少する。基質は、遷移状態を経て反応中間体、生成物へと変化する。基質と同様、生成物もポテンシャルエネルギーが極小の状態である。
具体的な例で示すと、ペプチド結合のカルボニル基のsp2炭素原子から伸びる結合は平面状の構造になっている。これが加水分解を受ける際には水酸化物イオンの求核攻撃を受けてsp3の正四面体構造の反応中間体となり、アミノ基が脱離することで再びsp2炭素に戻ることで平面構造のカルボニル基が再生し安定化する。この2段階の素反応のそれぞれに遷移状態がある。通常の有機反応においては、水酸化物イオンが求核攻撃する際の衝突エネルギーが内部ポテンシャルエネルギーを増大させ、ポテンシャルエネルギーが極大のsp3の正四面体構造へと変化させる原動力になっている。逆に水酸基以外の要因でペプチド結合の距離や角度が、遷移状態や反応中間体に近い形にあらかじめ変化させられていれば、水酸化物イオンが与える衝突エネルギーは少なくて済む。
基質が酵素と結合すると、酵素は誘導適合によりコンホメーションが変化させられるが、基質側のコンホメーションも酵素により変化させられ、その原子間距離や角度が遷移状態に近い形状に変形させられる。これが、酵素の活性化エネルギーが有機化学の触媒の活性化エネルギーよりも小さい理由のひとつになっている。
酵素と同じように分子構造を識別し、その分子と結合する生体物質に抗体がある。1986年、アメリカのトラモンタノらは、酵素と同じ働きをするように意図して製造した抗体が意図通りの酵素作用を示すことを発見し、抗体酵素 (abzyme) と名づけた。抗体酵素は、ある基質の遷移状態の原子距離・角度に近い物質(基質アナローグ)に対する抗体である。その抗体と元の基質とを反応させると、基質と基質アナローグとは分子構造が似ている為、基質もその抗体と結合する。その抗体に誘導適合する際には基質は基質アナローグに似た形状である遷移状態に近い形状に変形させられる。
酵素反応の調節機構[編集]
生体が酵素活性の大小を制御するには、酵素の量を制御する場合と、酵素の性質を変化させる場合とがある。それらは次のように分類される:[3]
- 酵素タンパク質の合成量制御による酵素量の増大
- 酵素タンパク質が他の生体分子と可逆的に作用することによる酵素活性の変化
- 酵素タンパク質が修飾されることによる酵素活性の変化
1 の調整は遺伝子の発現量の転写調節により実現する(詳しくはオペロンおよびラクトースオペロンを参照。ただし原核生物のみ)。例えば、細胞内のコレステロール量が減少すると、コレステロール代謝の律速段階であるHMG-CoAリダクターゼが遺伝子より翻訳生産され、コレステロールの生産量を増大させる(詳しくは記事 コレステロール#調節を参照のこと)。ただし、一度生産された酵素がタンパク質分解酵素(プロテアーゼ)により分解消失するまでには一定の寿命期間があることと、遺伝子からタンパク質が生産されるにはある程度時間が必要であることから、この調節には時間がかかる。
2 や 3 については酵素の質的な変化であり、1 の転写制御より素早い応答を示す。
2 や 3 の調節の例として、フィードバック阻害が挙げられる。一般に触媒反応の反応速度は基質濃度と生成物濃度により影響を受けるが、酵素反応の場合、ある複数の段階からなる代謝経路において、酵素の直接の基質あるいは生成物以外の代謝生成物が酵素の反応速度を制御する場面が良く見られる。特に、代謝生成物が過剰になったときに、生成物が何段階か上流過程の酵素反応を阻害することで産生を抑制する調節過程を、フィードバック阻害と呼ぶ。アロステリック効果などフィードバック阻害がかかる場合、生産物が過剰になると酵素活性が低減し、生産物が減ると酵素活性は復元する。
あるいは、細胞内キナーゼで酵素タンパク質がリン酸化されて酵素活性が発現する場合は、リン酸化された酵素が分解消失したり、他の酵素によりリン酸基の修飾が除去されるまでは酵素活性は維持される。また、消化酵素のトリプシンは、トリプシノーゲンとして膵臓から分泌されたあと、十二指腸表面に存在する酵素エンテロペプチダーゼ (EC 3.4.21.9) によりペプチド鎖の Lys6-Ile7 間を分解切断されてトリプシンとなり活性を発現する。また、熱ショックタンパク質を代表とする分子シャペロンは酵素の高次構造を変化させることで酵素を不活性型から活性型へと変化させる。
アロステリック効果
アロステリック効果は、生体内におけるフィードバック阻害の一例である(詳しくはアロステリック効果を参照)。
アロステリック効果アロステリック効果(アロステリックこうか)とは、タンパク質の機能が他の化合物(制御物質、エフェクター)によって調節されることを言う。主に酵素反応に関して用いられる用語であるが、近年、Gタンパク質共役受容体 (GPCR) を中心とする受容体タンパク質の活性化制御において、アロステリック効果を示す化学物質 (アロステリックモジュレーター) の存在が知られるようになってきた。 アロステリー(allostery、その形容詞がアロステリックallosteric)という言葉は、ギリシア語で「別の」を意味するallosと「形」を意味するstereosから来ている。これは、一般にアロステリックタンパク質のエフェクターが基質と大きく異なる構造をしていることによる。このことから、制御中心が活性中心から離れた場所にあると考えられたのである。 しかし下記のヘモグロビンにおける酸素分子のように、同じ分子がエフェクターかつ基質となる例もあり、アロステリック効果は一般にヘモグロビンのようなオリゴマー構造でモデル化することができる(「アロステリック制御のモデル」の項参照)。 このため、アロステリック効果は タンパク質と化合物が一対多の複合体を形成する際に、前の段階の複合体形成によって次以降の複合体形成反応が促進・抑制されること、あるいはその複合体による反応が加速・減速されること。 と拡張定義されることも多い。 アロステリック制御アロステリック効果により主に酵素や受容体などのタンパク質の機能が制御される現象をアロステリック制御と呼ぶ。 酵素の場合、酵素の活性中心以外の部分(アロステリック部位)に対してエフェクター分子(反応に関係する物質でもそうでなくてもよい。)が会合して酵素のコンフォメーションが変化し、酵素の触媒活性や複合体形成反応の平衡定数が増減することを表す。 酵素の活性を促進するエフェクターはアロステリック・アクティベーターと呼ばれ、逆にタンパク質の活性を抑制するエフェクターはアロステリック・インヒビターと呼ばれる。アロステリック制御はフィードバック調節の一つの例である。 受容体の場合、内因性アゴニストのアゴニスト活性を促進するアロステリック部位に結合するリガンドはポジティブアロステリックモジュレーター (Positive Allosteric Modulator, PAM) と呼ばれ、逆にアゴニスト活性を抑制するアロステリックリガンドはネガティブアロステリックモジュレーター (Negative Allosteric Modulator, NAM) と呼ばれる。アロステリック部位に結合するだけで内因性アゴニストの活性に影響を与えないリガンドはサイレントアロステリックモジュレーター (Silent Allosteric Modulator, SAM) 、もしくはニュートラルアロステリックリガンド (Neutral Allosteric Ligand, NAL) と呼ばれる。 例血液中のヘモグロビンは酸素と結合する鉄中心を持つヘムを四つ持ち、各々の酸素との結合には一定の平衡定数が存在する。しかし、ヘモグロビン中の一つのヘムが酸素と結合を作るとヘモグロビン全体の構造が変化し、他のヘムと酸素との結合が促進される。すなわち、酸素濃度の高い所では単独のヘムよりも効率的に酸素を取り入れることができる。一方で、細胞中のミオグロビンのそれぞれのヘムにはヘモグロビンのような協同効果は無いので、酸素との結合生成反応は酸素濃度に一次で比例するだけである。この結果、ヘモグロビンは酸素の多い肺では酸素を吸収し、酸素の少ない各細胞では酸素を放出することができるのである。 アロステリック制御のモデル多くのアロステリック効果はジャック・モノー、ワイマン、ジャン・ピエール・シャンジューの唱える協奏モデルと、モノー・ワイマン・シャンジューモデルとダニエル・コシュランド、ネメシー、フィルマーの提唱する逐次モデルの両方で説明できる。どちらの説でも酵素サブユニットは緊張(T状態)か弛緩(R状態)のどちらかの状態にあると仮定し、弛緩状態のサブユニットは緊張状態のサブユニットよりも基質に結合しやすいとしている。二つのモデルは、サブユニット同士の関係と、両方の状態に至る前の状態に関する仮定の面で異なっている。 協奏モデルアロステリックに関する協奏モデルは対称モデルともモノー・ワイマン・シャンジュー (MWC) モデルとも呼ばれるが、一つのサブユニットの構造変化が他のサブユニットに影響を与えると仮定している。つまり、全てのサブユニットが同じコンフォメーションを取る。このモデルはリガンドがなくても成り立ち、T状態とR状態のコンフォメーションが均衡を保っている。一個のリガンド(もしくはアロステリックエフェクター)がアロステリック部位に結合すると、均衡はR状態もしくはT状態に移行する。 逐次モデルアロステリック制御の逐次モデルでは、一つのサブユニットのコンフォメーション変化が他のサブユニットに同様の変化を引き起こすとは考えない。つまり全てのサブユニットが同じコンフォメーションをとっている必要はない。さらに逐次モデルでは、基質分子が誘導適合モデルによって結合するとしている。一般的には、サブユニットがランダムに基質分子と衝突した時、活性中心が基質を包み込まなければならない。この誘導適合はサブユニットをT状態からR状態に移行させるが、近接サブユニットの構造を変化させることはない。その代わり、一つのサブユニットに基質が結合すると他のサブユニットの結合部位も基質に結合しやすいように徐々に構造を変えていく。要約すると、
アロステリック促進酸素分子がヘモグロビンに結合する時のように、アロステリック促進はリガンドの結合が基質分子と他の結合サイトの反応性を高める現象である。ヘモグロビンの例では、酸素は基質であると同時にエフェクターとして、効率的に働いている。アロステリックサイトは、隣のサブユニットの結合部位である。一つのサブユニットに酸素が結合すると、構造が変化し、残りの結合部位の酸素親和性を高める。 アロステリック抑制アロステリック抑制は、リガンドの結合によって結合部位の基質親和性が低下する現象である。例としては、2,3-ビスホスホグリセリン酸がヘモグロビンのアロステリック部位に結合すると、他の全てのサブユニットの酸素への親和性が低下する。 代謝系の生産物が、その系の中間反応を触媒する酵素の活性を抑制する場合、負のフィードバック制御の生体内における例であるとみなせるため、フィードバック阻害と呼ばれる。 エフェクターのタイプ多くのアロステリックタンパク質は自身の基質によって調節される。これらはホモトロピックアロステリック分子と呼ばれ、多くはアロステリック促進を示す。非基質の制御分子はヘテロトロピックアロステリック分子と呼ばれ、促進作用を示すものも抑制作用を示すものもある。自身の基質と非基質分子の両方で調節されるアロステリックタンパク質もある。このようなタンパク質はホモトロピック作用もヘテロトロピック作用も受ける。 関連項目
<img src="//ja.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1x1" alt="" title="" width="1" height="1" style="border: none; position: absolute;" /> 「https://ja.wikipedia.org/w/index.php?title=アロステリック効果&oldid=62943660」から取得 カテゴリ: |
||||||
アロステリック酵素は活性中心近傍の基質結合部位とは異なる場所に低分子物質を結合させ、その活性を変化させる。そうしたアロステリック効果を誘導する低分子物質をアロステリックイフェクターと呼ぶ。
例えば、アスパラギン酸からリジンを合成する反応系では、最終産物のリジンがアロステリックイフェクターとなる。リジンが少量であるときは、アスパラギン酸キナーゼは盛んに触媒作用を発揮するが、リジン過剰になるとアスパラギン酸キナーゼのリジン結合部位にリシンが結合し、アスパラギン酸キナーゼの活性が低下する。
|
ピックス |
|
|
タイプ |
EC1 酸化還元酵素 EC2 転移酵素 EC3 加水分解酵素 EC4 リアーゼ EC5 異性化酵素 EC6 リガーゼ |
逆にアロステリックイフェクターが正の方向に作用するケースもあるが、反応最終生産物の関与するアロステリック効果はほとんどの場合に活性を低下させる。
出典
- ^ 一島英治 『酵素の化学』 朝倉書店、1995年。ISBN 4-254-14555-1
- ^ 『新生化学ガイドブック』 南江堂、1969年、89頁。
- ^ 代謝調節 『理化学辞典』 第5版、岩波書店。
関連項目
酵素反応速度論
|
|

大腸菌のジヒドロ葉酸還元酵素。活性部位に2つの基質ジヒドロ葉酸 (右) とNADPH (左) が結合している。蛋白質はリボンダイアグラムで示されており、αヘリックスは赤、ベータシートは黄、ループは青に着色されている。7DFRから作成。
酵素反応速度論 (こうそはんのうそくどろん) とは酵素によって触媒される化学反応を反応速度の面から研究する学問。酵素の反応速度論を研究することで、酵素反応の機構、代謝における役割、活性調節の仕組み、薬物や毒が酵素をどう阻害するかといったことを明らかにできる。
酵素は通常蛋白質分子であり、他の分子 (酵素の基質という) に作用する。基質は、酵素の活性部位に結合し、段階的に生成物へと変化を遂げる。この過程は、反応機構と呼ばれる。反応機構は、単一基質機構と、複数基質機構に分類できる。
一つの基質としか結合しない酵素、例えばトリオースリン酸イソメラーゼの研究では酵素が基質と結合する際の解離定数や回転率の測定を目指す。
酵素が複数の基質と結合する場合、例えばジヒドロ葉酸還元酵素 (右図) では、基質が結合し、生成物が解離する順序を明らかにすることもできる。1つの基質と結合し、複数の生成物を放出する酵素の例としてプロテアーゼ が挙げられる。この酵素は1つの基質蛋白質を切断して、2つのポリペプチドにする。2つの基質を1つに結合する酵素もある。DNAポリメラーゼはヌクレオチドをDNAに結合する。これらの酵素の反応機構は複雑で何段階にも及ぶことが多いが、通常律速段階があって、これが全体の反応速度を決定する。律速段階は化学反応であったり、(生成物が酵素から離れる際など) 酵素や基質のコンフォメーションの変化であったりする。
酵素の立体構造が分かると、速度論的な情報の解釈に有利である。例えば構造から触媒過程で基質や生成物がどのように結合しているか、反応中にどう変化するかが分かる。また、特定のアミノ酸残基が反応機構で果たす役割が分かることもある。酵素によっては反応中に構造が大きく変化するものがあるが、このような場合は酵素の構造を触媒を受けない基質類似体が結合した場合と、結合していない場合それぞれで決定しておくとよい。
生体で触媒として働くのは蛋白質でできた酵素だけではない。RNAによる触媒、つまりリボソームのようなリボザイムは、RNAスプライシングや翻訳といった多くの過程で不可欠である。リボザイムと酵素の主要な違いは、RNAのほうが触媒できる反応が限られているということだ。もっとも、RNAによる触媒の機構も、蛋白質酵素の場合と同じ方法で解析し、分類することができる。
総論
|
|
.svg)
反応速度は基質 (substrate) 濃度に合わせて上昇するが、基質濃度が十分高くなると飽和する。
酵素によって触媒される反応でも、その反応物と生成物は触媒を受けていない場合と変わらない。他の触媒と同じく、酵素も反応物と生成物の間の化学平衡を動かすことはない[1]。ただし、触媒されていない反応と違って、酵素触媒下の反応は反応速度に飽和点を持つ。ある酵素濃度のもとで基質濃度が相対的に低いと、反応速度は基質濃度に比例して上昇する。酵素分子は自由に反応を触媒でき、基質濃度が上がれば基質と酵素が出会う頻度も上昇するからだ。ところが基質濃度がある程度高くなると、反応速度は理論的上限値に漸近する。酵素の活性部位は、ほとんど全て基質と結合してしまい、反応速度は酵素固有の回転率によって決まるようになる。この両極端のちょうど中間にあたる基質濃度は、KMで表される。
酵素の速度論的特徴の中で特に重要なのは特定の基質に対して酵素が飽和するのはいつかということと、その時の最大反応速度がどれだけかということである。これらの特徴から、酵素が細胞の中でどんな働きをしており条件が変化したときに酵素がどう反応するかを推察できる。
酵素の試験
|
|

酵素反応の進行を図にしたもの。初期速度区間 "initial rate period" と示された部分の傾きは、初期速度 v を表す。 ミカエリス-メンテンの式は基質濃度を変化させたときにこの傾きがどう変わるかを表現する。
酵素の試験 (アッセイ) とは、実験室で酵素反応の速度を測ることである。酵素は反応を触媒する過程で消費されることはないため、実際に測るのは基質か生成物の濃度変化である。測定方法にはいろいろある。紫外・可視・近赤外分光法では、生成物と反応物の吸光度の違いを測る。放射能分析では生成物が徐々にできてくるのを放射性元素の取り込みや放出によって測る。分光法は、反応速度を連続的に測定できるので便利である。放射能分析では、サンプルを取り出して測定する必要がある (非連続的分析である) が、多くの場合非常に鋭敏で、ごくわずかな酵素活性でも測定できる[2]。よく似た方法として質量分析器を使う手もあり、基質が生成物へ変化する過程で安定同位体が取り込まれたり放出されるのを観測する。
最も敏感な酵素測定法はレーザを使う方法である。顕微鏡下で酵素1分子にレーザの焦点を合わせ、反応を触媒する過程での変化を観察する。反応中に補因子の蛍光が変化するのを測定したり、蛋白質の一部分を蛍光色素でラベルして触媒中の動きを調べる[3]。こうした実験から酵素単一分子の速度論やダイナミクス (反応中の動き) について新しい知見が得られつつある。従来の方法は数百万にも及ぶ多数の酵素分子の挙動を平均的したものを観察していた[4][5]。
酵素測定における典型的なグラフが左に示してある。反応開始時点では初期速度にしたがって線形に (時間に比例して) 生成物ができる。時間が経つ (グラフの右) と反応速度は低下する。基質が消費され生成物が蓄積するからだ。初期速度で反応が進む時間の長さは、測定条件によって変わり、数ミリ秒のこともあれば、数時間に及ぶこともある。通常の測定ではこの期間が約1分になるようにしておくと実験が容易である。ただし、液体を急速に混ぜ合わせる装置を使えば初期速度の段階が1秒に満たない反応でも測定できる。これをストップフロー法という[6]。これらの高速な測定技術は、後述するように定常状態に達する前の速度を調べるのに不可欠である。
酵素反応速度論の実験では反応のこの初期部分、つまり生成物が時間に比例してできてくる線形部分に注目することが多い。しかし、反応全体を測定してそのデータを非線形の速度方程式に当てはめることもできる[7]。このような測定をprogress-curve 解析という。この方法は初期速度が速すぎて正確に測定できないときに有効である。
単一基質反応
単一基質機構を持つ酵素としてはトリオースリン酸イソメラーゼや二ホスホグリセリン酸ムターゼのようなイソメラーゼや、アデニル酸シクラーゼやRNAリアーゼであるハンマーヘッドリボザイムのような分子内リアーゼがある。ただし基質が一つしかない酵素でも反応機構は単一基質機構でないことがある。カタラーゼがその例で、まず基質である過酸化水素の1つ目の分子と反応して酵素自身が酸化され、続いて2つ目の分子によって還元される。確かに基質は1種類だが修飾された酵素が中間体として存在している以上、カタラーゼの反応機構はピンポン機構というべきである。これについては複数基質反応として後述する。
ミカエリス-メンテン速度論
|
|
|
|
酵素反応の飽和曲線。基質濃度と反応速度の関係を示す。 |
酵素反応の単一基質機構。 k1、 k-1 、k2 は、各段階の速度定数である。詳細は「ミカエリス・メンテン式」を参照 |


酵素によって触媒される反応は飽和を示すので、反応速度は基質を増やしてもそれに比例して線形に増えるわけではない。反応の初期速度を基質濃度 ([S]で表す) を変えながら測定すると、反応速度 (v) は右に示すように、[S] にあわせて上昇する。しかし[S] がさらに増えると酵素は基質で飽和し、反応速度は酵素の最大反応速度 Vmaxに達する。
単一基質反応におけるミカエリス-メンテンのモデルを右に示した。まず酵素 E と基質 S が反応して、酵素基質複合体 ES を作る二分子反応が起こる。単分子反応 で表される触媒反応の機構は実際には複雑かもしれないが、通常1つの律速段階があって、触媒作用を速度定数k2を持つ1つの触媒反応で表現できる。
(式 1).
k2は、kcatや回転数とも呼ばれ、酵素が1秒に行える反応回数の上限を示す。
基質濃度 [S] が小さい場合、酵素は遊離型 E と基質酵素複合体 ES の平衡状態にある。[S] を増やすと[E] が減って [ES] が増え、平衡が右に傾く。反応速度は [ES] によって決まるため、[S] のわずかな変化でも反応速度が大きく変わる。しかし、[S] が非常に大きくなると、酵素は完全に基質で飽和し、全て基質酵素複合体 ES となる。こうなると、反応速度 (v≈k2[E]tot=Vmax) は、[S]が少し変化したくらいでは変わらない。ここで、[E]totは、酵素の全濃度であり、
飽和条件での [ES] 濃度にほぼ等しい。
ミカエリス-メンテンの式[8]は、反応速度 v が、酵素基質結合の平衡や速度定数 k2 とどう関係するかを示す式である。レオノール・ミカエリスとモード・レオノーラ・メンテン は、 k2 が k-1 よりもずっと小さいとき (平衡の仮定)、次の式を導いた[9]。
(式 2)
この式は、単一基質機構をもつ酵素ほとんどの速度論の基礎となる。
ミカエリス定数 Km は、酵素の反応速度が Vmax の半分になるときの基質濃度として定義されている。このことはミカエリス-メンテン式で[S] = Km を代入してみれば確認できる。反応の律速段階が基質の解離よりもずっと遅い場合 (k2 << k-1)、ミカエリス定数 Km は複合体 ES の解離係数に概ね等しい。ただし、このような状況は比較的まれである。
多くの状況では、k2 > k-1 であり、Briggs-Haldane 状態と呼ばれる[10]。定常状態近似から分かるように、ミカエリス-メンテン式はこれらの状況でも成立する。初期速度の段階では反応速度はほとんど一定であり、[ES] も変化しないことが分かる (式1)。
よって、[ES] は下の式であたえられる。
ここでミカエリス定数 Km の定義は、
([E] は遊離の酵素濃度)。まとめると、反応速度 v の一般式は再びミカエリス-メンテン式に戻る。
特異度定数 は、酵素が基質を生成物に変換する際の効率を示す。ミカエリス定数の定義から、ミカエリス-メンテンの式は次のような形にも書ける。
ここで[E] は遊離の酵素濃度である。つまり、特異度定数とは遊離の酵素が遊離の基質と結合して生成物をつくる際の実質的な2次の速度式である。特異度定数は溶液中で酵素と基質が出会う頻度によって制限されるが、その値は1010 M−1 s−1にも及ぶ[11]。驚くべきことだが、この最大速度は基質や酵素の大きさとはほとんど関係ない[12]。2つの基質があったとき、その特異度係数の違いは酵素がそれぞれの基質を変換する際の効率の違いを定量的に表す。基質濃度 [S] が小さいとき ([S] << Km)、ミカエリス-メンテン式のグラフの傾きから、特異度定数を知ることもできる。
ミカエリス-メンテン式の線形プロット
|
|

ラインウィーバー=バークプロット、別名二重逆数プロットで、速度論的データを示したもの。切片と傾きの意味も表示してある
バージニア大学の、ミカエリス-メンテン速度論のチュートリアル (英語) で[注 1]、速度論的パラメータが変わったときの酵素の挙動を試してみることができる。
v と [S] を縦横に取ってグラフを描くと、直線にはならない。[S] が小さいうちは線形だが[S] が大きい部分では飽和し、グラフが曲がってくるのだ。コンピュータで非線形回帰分析ができるようになる前はこの非線形性のせいでKm と Vmax を正確に読み取ることは難しかった。そのため、ラインウィーバー=バークプロット、イーディー=ホフステー図、ヘインズ=ウルフプロット といった線形化手法が編みだされた。これらの表現法は、データを表示するには有用だが、速度係数を求めるためにはもはや使われていない。非線形回帰によってより正確に係数を求めるソフトウェアがあるためだ[13]。
ラインウィーバー=バークプロット(別名:二重逆数プロット)は速度論的データを示す際によく使われる。これはミカエリス-メンテン式の両辺の逆数を取ると得られる。右に示したようにミカエリス-メンテン式の線形化であり、y = mx + c 型の直線となる。y 切片は 1/Vmax に等しく、x切片は -1/Km を表す。
もちろん、 1/[S] が負となるような部分の数値を測定することはできない。1/[S] = 0 である下限 (y切片) は基質濃度無限大にあたり、右に示すように 1/v=1/Vmax である。また、x切片は、正の濃度における実験データから外挿したものである。しかもラインウィーバー=バークプロットは、基質濃度が低い条件で測定したデータを過大評価しており、Vmax とKmを不正確にする恐れがある[14]。より正確な線形プロットは、イーディー=ホフステー図である。この図ではv を、v/[S] に対してプロットする。3つ目の線形プロットは、ヘインズ・ウルフプロットであり、[S]/v を [S] に対してプロットする。どの場合でも、データを正規化することで、実験の量を減らし、結果の信頼性を改善することができるし、視覚的または数値的な解析にも適している[15]。
速度定数の実際的な意義
酵素の速度論を研究することは二つの基本的な理由から重要である。まず、酵素がどう働くかを考えるのに役立つ。また、生体内で酵素が示す挙動を予測するのにも役立つ。上のように定義された速度定数 Km と Vmax は、複数の酵素が共同して代謝を制御する仕組みを理解する上で不可欠である。
このような制御を理解することは、単純な系においてさえ容易ではない。例えば、オキサロ酢酸は、ミトコンドリアの中でリンゴ酸デヒドロゲナーゼによって生成される。その後、リンゴ酸シンターゼ、ホスホエノールピルビン酸カルボキシラーゼ、アスパラギン酸アミノトランスフェラーゼなどによって消費され、それぞれクエン酸回路、糖新生、アスパラギン酸生合成へと進む。それぞれの経路にどれだけのオキサロ酢酸が進むかを予測するには、オキサロ酢酸の濃度だけでなく各酵素の濃度と速度論的係数の情報が不可欠である。代謝経路の動態を予想することの究極の目的は酵素の速度論と遺伝子発現の莫大なデータを1個体全体の数学的モデルとしてまとめあげることにある。この目標は真核生物に関しては、まだまだ先が長いが、バクテリアに関しては既にEscherichia coliのモデルが作られて試験中である[16][17]。
複数基質反応
複数基質反応の速度方程式は、基質結合の仕方や順序を記述する複雑なものとなる。このような反応を解析する際、1つ目の基質 A の濃度を一定にして2つ目の基質 B の濃度だけを変えると簡単である。この場合、単一基質酵素と同じ挙動になり、v を [S] に対してプロットすると、基質 B に対する見かけ上の定数 Km と Vmax が得られる。いくつかの A の濃度についてこの測定をすれば、反応機構を考察するのに役立つデータが得られる。2つの基質 A と B を生成物 P と Q に変化させる酵素の場合は、反応機構として三重複合体機構とピンポン機構の二つがある。
三重複合体機構
|
|
|
順不同型 (random-order) 三重複合体機構。反応経路を線で示し、その下に 基質 A, B や 生成物 P, Q を含んだ酵素中間体が示してある。 |

このタイプの酵素では基質が2つとも同時に酵素に結合し、E-A-B という三重の複合体を作る。基質結合の順序は、決まっていない場合 (random-order) と決まっている場合 (ordered) がある。三重複合体機構を持つ酵素について、A の濃度を固定して B の濃度 [S] に対する v のグラフをラインウィーバー=バークプロットで描く。A の濃度を変えてグラフを何本か描くと、これらの直線は一点で交わる。
三重複合体機構を持つ酵素の例としては、グルタチオン-S-トランスフェラーゼ[18]、ジヒドロ葉酸還元酵素[19]、DNAポリメラーゼ[20]が挙げられる。次のリンク先 (英語) は、ジヒドロ葉酸還元酵素やDNAポリメラーゼの反応機構を短いアニメーションで紹介している[注 2][注 3]。
ピンポン機構
|
酵素反応におけるピンポン機構。中間体は、基質 A, B と生成物 P, Q を含む。 |
右に示したように、ピンポン機構を示す酵素は、E と修飾型 E* という2つの状態を持つ。修飾型 E* は 反応中間体 と呼ばれる。この機構では、基質 A が結合すると酵素は E* になる。例えば、反応中心に基質の一部が転移し、基質の残りの部分が解離する。1つ目の基質が離れてはじめて2つ目の基質が修飾型酵素 E* に結合し、反応できる。反応した酵素は非修飾型 E に戻る。A の濃度を固定して、B の濃度を変えながら [S] に対する v のグラフをラインウィーバー=バークプロットで描く。いくつかの A 濃度についてこれを行うと、平行線となる。
ピンポン機構を示す酵素としては、チオレドキシンペルオキシダーゼのような酸化還元酵素[21]、アシルノイラミン酸シチジル転移酵素のような転移酵素[22]、トリプシンやキモトリプシン のようなセリンプロテアーゼ[23]がある。セリンプロテアーゼは非常に普遍的かつ多様な酵素群であり、消化酵素 (トリプシン、キモトリプシン、エラスターゼ) や血液凝固カスケードの酵素の一部も含まれる。セリンプロテアーゼの場合、E* 中間体はアシル-酵素複合体であり、基質蛋白質のペプチド結合に活性部位のセリン残基が求核付加してできる[注 4]。
非ミカエリス-メンテン速度論
|
シグモイド型を示す酵素の飽和曲線 |

v を [S] に対してプロットした時 シグモイド曲線 となる酵素がある。これは、活性部位への基質結合が協調的であることを示す場合が多い。つまり、1つの基質結合が、続く基質結合に影響するのである。この現象は、複数の活性中心が相互作用しているような多量体酵素でよく見られる。[24] 協調の機構はヘモグロビンの場合と似ており、基質が1つの活性部位に結合すると、他の活性部位の基質への親和性が変化するのである。正の協調は、1つ目の基質結合が他の活性部位の基質親和性を高める場合に起こる。負の協調は、1つ目の基質結合が他の活性部位の親和性を下げる場合に起きる。
アロステリック酵素の例としては、負の協調を示す哺乳類のチロシンt-RNA合成酵素[25]、正の協調を示すバクテリアの アスパラギン酸トランスカルバモイラーゼ[26]やホスホフルクトキナーゼ[27]が挙げられる。 協調現象は驚くほど普遍的であり、基質濃度の変化にあわせて酵素の反応を変えるのに役立っている。正の協調を示す酵素は [S] に敏感に反応し、基質濃度のわずかな変化で大きな活性変化を見せる。負の協調では、逆に、[S] が少し変化したぐらいでは活性があまり変化しない。
Hill 方程式 は[28]非ミカエリス-メンテン型の速度論において、協調の程度を定量的に表すのによく使われる。この式から得られる Hill 定数 n は、基質の1つの活性部位への結合が他の活性部位での基質結合にどう影響するかを表す。1未満の値は負の協調を表し、1を越える値は正の協調を示す。
前定常状態速度論
|
定常状態に達する前の反応のグラフ。酵素反応の burst 期を示している。 |

酵素を基質と混合した直後には、生成物はまだできていないし、反応中間体も存在しない。この後数ミリ秒間の反応を扱う学問を、前定常状態速度論という。つまり、前定常状態速度論では、定常状態以前の段階での酵素基質中間体 (ES や E*) の生成と消費を扱う。
この手法が最初に用いられたのはキモトリプシンによる加水分解反応の研究であった。反応機構の調査では、中間体を検出することが動かぬ証拠となることが多い。例えば、前述のピンポン機構では、高速な速度論的測定によって生成物 P の放出や修飾された酵素中間体 E* の生成を検出できる。キモトリプシンの場合、活性部位にある求核性セリンが基質を攻撃することで、アシル酵素中間体ができる。
右図では、酵素から E* が反応開始後数秒のうちに急速にできる。定常状態に達すると速度は低下する。反応が爆発的に進むこの期間 (burst 期) は、酵素の単一回転に相当する。よって、この間に放出される生成物は (グラフのy切片で表されているが)、その測定において活性を持っていた酵素の量をも表す[29]。
化学的機構
酵素の速度論を研究する重要な目的の1つが、酵素反応の機構を化学的に説明することである。基質が生成物に至る化学反応の各段階を明らかにするのである。上に述べたような速度論的手法によって、反応中間体がどんな速度で生成し、相互変換するかを明らかにできるが、各中間体が何なのかを確実に決定することはできない。
反応液の条件を変えたり、酵素に変異を加えたり、少し異なる基質を使って速度論的解析を行うと、機構の化学的側面が分かってくることが多い。反応の律速段階や中間体が明らかになるからである。例えば、水素原子との共有結合の切断はよく律速段階となっている。考えられる水素転移のうちどれが律速段階であるかは、水素原子を1つずつ安定同位体 (重水素) に置換することで調べられる。律速段階にかかわる水素原子が置換されると、一次同位体効果によって反応速度が変化する。重水素への結合は通常の水素への結合よりも切れにくいからである[30]。13C/12C や 18O/16O など他の原子についても同じような効果を観測することができるが、水素の場合ほど顕著ではない[31]。
同位体を使えば、基質分子の各部分が生成物分子のどこに対応するかを調べることもできる。例えば、最終産物中の酸素原子の由来を明らかにするのは難しいことが多い。水から来たのかもしれないし、基質から来たかもしれないからだ。これをはっきりさせるには、反応に関与する分子に含まれる酸素原子を、1つ1つ安定同位体 18O で置換して、生成物中の同位体を調べればよい[32]。酵素の化学的機構を調べるには、溶液の pH を変えながら反応速度や同位体効果を調べるのもよい[33]。金属イオンなどの補因子を変えたり[34]、保存されているアミノ酸残基を選択的に変異させたり、基質アナログの存在下で酵素の挙動を調べるのも有用である[35]。
酵素の阻害
|
|

可逆的阻害剤の速度論的特徴
酵素阻害剤とは、酵素の活性を弱めたり、完全に失わせる分子である。阻害剤には、可逆的 (阻害剤を取り除けば酵素活性が元に戻る) なものと不可逆的 (酵素を永久的に失活させる) なものとがある。
可逆的阻害剤
可逆的阻害剤は、Km と Vmax に及ぼす影響の違いによって、競合的、不競合的、非競合的、混合型に分類される。これは、阻害剤が結合する相手が、酵素 E なのか、酵素基質複合体 ES なのか、両方なのかによって決まる (右図および、下表参照)。阻害剤がどのタイプにあたるのかは、阻害剤濃度を変えながら酵素の反応速度を測れば分かる。阻害剤濃度ごとに ラインウィーバー=バークプロット や イーディー=ホフステー図 を描くと[36]、阻害剤の種類によってグラフの変化のしかたが異なる。記述を簡単にするために、
and
とおく。ここでKi と K'i はそれぞれ、酵素または基質酵素複合体に対する阻害剤の解離定数である。可逆的阻害剤があると、酵素の見かけ上の Km とVmax は、それぞれ (α/α')Km と (1/α')Vmax に低下する。
|
阻害剤の種類 |
見かけ上の Km |
見かけ上の Vmax |
||
|
Ki only |
() |
競合的 |
|
|
|
Ki' only |
() |
不競合的 |
|
|
|
Ki = Ki' |
() |
非競合的 |
|
|
|
Ki ≠ Ki' |
() |
混合型 |
|
|
非線形回帰 によって酵素の速度論的データを上記の速度方程式にフィットすれば[37]、解離定数 Ki と K'i を正確に算出できる。
不可逆的阻害剤
阻害剤には、酵素を不可逆的に阻害するものもある。不可逆的阻害剤の多くは、活性部位の残基と共有結合を作って酵素を修飾してしまい、自殺基質と呼ばれる。このような反応は指数的減衰を示し、通常飽和がある。飽和未満では阻害剤との反応は一次反応の速度論を示す。
触媒の機構
|
|

反応座標に対するエネルギー変化の図。酵素は遷移状態を安定化させる。
酵素と基質の相互作用を表すモデルとしてよく使われるのが induced-fit モデルである[38]。このモデルでは、酵素-基質相互作用は始めのうちこそ弱いが、この相互作用によって酵素の立体配座 (コンフォメーション) がまもなく変化し、基質との結合が強くなると考える。また、立体配座の変化のため、活性部位の触媒に関わる残基が基質中の変化を受ける結合に近づく[39]。結合が成立すると、何らかの機構で反応の遷移状態のエネルギーが低下する。つまり、反応の別経路ができる。触媒の機構としては、結合のひずみによる触媒、反応部位同士が近接して適切な位置関係にあることによる効果、活性部位からのプロトン授受による触媒、共有結合を伴う触媒、量子トンネル効果などがある[40]。
酵素の速度論的考察だけでは、触媒の種類を特定することはできない。しかし、速度論的データから可能性を絞りこんだのち、別の手段で確認することができる。例えば、burst 期前定常状態を示すピンポン機構型酵素では共有結合が触媒過程で重要だろうと推測できる。また、pH を変えたときにVmax が大きく変わるが Km はあまり変化しない酵素の場合、活性部位が特定のイオン化状態にあることが触媒に重要なのだろうと言える。
注釈
リンク先は英語のサイトである。
- ^ Link: Interactive Michaelis–Menten kinetics tutorial (Java required)
- ^ Link: dihydrofolate reductase mechanism (Gif)
- ^ Link: DNA polymerase mechanism (Gif)
- ^ Link: Chymotrypsin mechanism (Flash required)
参考文献
1. ^ Ebbing, D.D. General chemistry (4th edition) Houghton Mifflin Co. 1993, ISBN 0-395-63696-5
2. ^ Eisenthal R. Danson M.J. (Eds), Enzyme Assays: A Practical Approach. Oxford University Press (2002) ISBN 0-19-963820-9
3. ^ Xie XS, Lu HP. Single-molecule enzymology. J Biol Chem. 1999 Jun 4;274(23):15967-70. PMID 10347141
4. ^ Lu H (2004). “Single-molecule spectroscopy studies of conformational change dynamics in enzymatic reactions”. Current pharmaceutical biotechnology 5 (3): 261–9. doi:10.2174/1389201043376887. PMID 15180547.
5. ^ Schnell J, Dyson H, Wright P (2004). “Structure, dynamics, and catalytic function of dihydrofolate reductase”. Annual review of biophysics and biomolecular structure 33: 119–40. doi:10.1146/annurev.biophys.33.110502.133613. PMID 15139807.
6. ^ Gibson Q.H. Rapid mixing: Stopped flow Methods in Enzymology, (1969) 16:187–228
7. ^ Duggleby, R.G. Analysis of enzyme progress curves by non-linear regression. Methods in Enzymology, (1995) 249:61–90.
8. ^ Michaelis L. and Menten M.L. Kinetik der Invertinwirkung Biochem. Z. 1913; 49:333–369 English translation Accessed 6 April 2007
9. ^ Cha S (1968). “A simple method for derivation of rate equations for enzyme-catalyzed reactions under the rapid equilibrium assumption or combined assumptions of equilibrium and steady state”. J. Biol. Chem. 243 (4): 820–5. PMID 5638598.
10. ^ Briggs GE, Haldane JB. A Note on the Kinetics of Enzyme Action. Biochem J. 1925;19(2):338-9. PMID 16743508
11. ^ Stroppolo ME, Falconi M, Caccuri AM, Desideri A (2001). “Superefficient enzymes”. Cell. Mol. Life Sci. 58 (10): 1451–60. doi:10.1007/PL00000788. PMID 11693526.
12. ^ Davis ME, Madura JD, Sines J, Luty BA, Allison SA, McCammon JA (1991). “Diffusion-controlled enzymatic reactions”. Meth. Enzymol. 202: 473–97. PMID 1784185.
13. ^ Jones ME (1992). “Analysis of algebraic weighted least-squares estimators for enzyme parameters”. Biochem. J. 288 ( Pt 2): 533–8. PMID 1463456.
14. ^ Tseng SJ, Hsu JP. A comparison of the parameter estimating procedures for the Michaelis–Menten model. J Theor Biol. 1990 Aug 23;145(4):457–64. PMID 2246896
15. ^ Bravo, I.G., Busto, F., De Arriaga, D., Ferrero, M. A., Rodríguez-Aparicio, L. B., Martínez-Blanco H., Reglero, A. A normalised plot as a novel and time-saving tool in complex enzyme kinetic analysis Biochem. J. (2001). 358, 573–583. PMID 11577687
16. ^ Almaas E, Kovacs B, Vicsek T, Oltvai ZN, Barabasi AL. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature. 2004 Feb 26;427(6977):839-43. PMID 14985762
17. ^ Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 2003;4(9):R54. PMID 12952533
18. ^ Dirr H, Reinemer P, Huber R. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur J Biochem. 1994 Mar 15;220(3):645-61. PMID 8143720
19. ^ Stone SR, Morrison JF. Dihydrofolate reductase from Escherichia coli: the kinetic mechanism with NADPH and reduced acetylpyridine adenine dinucleotide phosphate as substrates. Biochemistry. 1988 Jul 26;27(15):5493–9. PMID 3052577
20. ^ Fisher PA. Enzymologic mechanism of replicative DNA polymerases in higher eukaryotes. Prog Nucleic Acid Res Mol Biol. 1994;47:371-97. PMID 8016325
21. ^ Akerman SE, Muller S. 2-Cys peroxiredoxin PfTrx-Px1 is involved in the antioxidant defence of Plasmodium falciparum. Mol Biochem Parasitol. 2003 Aug 31;130(2):75-81. PMID 12946843
22. ^ Bravo, I.G., Barrallo, S., Ferrero, M. A., Rodríguez-Aparicio, L. B., Martínez-Blanco H., Reglero, A. “Kinetic properties of the Acylneuraminate Cytidylytransferase from Pasteurella haemolytica A2”. Biochem. J. (2001) 358, 585-598. [1]
23. ^ Kraut J. Serine proteases: structure and mechanism of catalysis. Annu Rev Biochem. 1977;46:331-58. PMID 332063
24. ^ Ricard J, Cornish-Bowden A. Co-operative and allosteric enzymes: 20 years on. Eur J Biochem. 1987 Jul 15;166(2):255-72. PMID 3301336
25. ^ Ward WH, Fersht AR., Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity. Biochemistry. 1988 Jul 26;27(15):5525–30. PMID 3179266
26. ^ Helmstaedt K, Krappmann S, Braus GH., Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase. Microbiol. Mol. Biol. Rev. 2001 Sep;65(3):404-21 PMID 11528003
27. ^ Schirmer T, Evans PR., Structural basis of the allosteric behaviour of phosphofructokinase. Nature. 1990 Jan 11;343(6254):140-5. PMID 2136935
28. ^ Hill, A. V. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. (Lond.), 1910 40, iv-vii.
29. ^ Bender ML, Begue-Canton ML, Blakeley RL, Brubacher LJ, Feder J, Gunter CR, Kezdy FJ, Killheffer JV Jr, Marshall TH, Miller CG, Roeske RW, Stoops JK. The Determination of the Concentration of Hydrolytic Enzyme Solutions : a-Chymotrypsin, Trypsin, Papain, Elastase, Subtilisin, and Acetylcholinesterase. J Am Chem Soc. 1966 Dec 20;88(24):5890-913. PMID 5980876
30. ^ Cleland WW. The use of isotope effects to determine enzyme mechanisms. Arch Biochem Biophys. 2005 Jan 1;433(1):2–12. PMID 15581561
31. ^ Northrop D (1981). “The expression of isotope effects on enzyme-catalyzed reactions”. Annu. Rev. Biochem. 50: 103–31. doi:10.1146/annurev.bi.50.070181.000535. PMID 7023356.
32. ^ Baillie T, Rettenmeier A (1986). “Drug biotransformation: mechanistic studies with stable isotopes”. Journal of clinical pharmacology 26 (6): 448–51. PMID 3734135.
33. ^ Cleland WW. Use of isotope effects to elucidate enzyme mechanisms. CRC Crit Rev Biochem. 1982;13(4):385–428. PMID 6759038
34. ^ Christianson DW, Cox JD. Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annu Rev Biochem. 1999;68:33–57. PMID 10872443
35. ^ Kraut D, Carroll K, Herschlag D (2003). “Challenges in enzyme mechanism and energetics”. Annu. Rev. Biochem. 72: 517–71. doi:10.1146/annurev.biochem.72.121801.161617. PMID 12704087.
36. ^ Tseng SJ, Hsu JP. A comparison of the parameter estimating procedures for the Michaelis–Menten model. J Theor Biol. 1990 Aug 23;145(4):457–64. PMID 2246896
37. ^ Leatherbarrow RJ. Using linear and non-linear regression to fit biochemical data. Trends Biochem Sci. 1990 Dec;15(12):455–8. PMID 2077683
38. ^ Koshland DE, Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. U.S.A. 1958 Feb;44(2):98–104. PMID 16590179
39. ^ Hammes G (2002). “Multiple conformational changes in enzyme catalysis”. Biochemistry 41 (26): 8221–8. doi:10.1021/bi0260839. PMID 12081470.
40. ^ Sutcliffe M, Scrutton N (2002). “A new conceptual framework for enzyme catalysis. Hydrogen tunnelling coupled to enzyme dynamics in flavoprotein and quinoprotein enzymes”. Eur. J. Biochem. 269 (13): 3096–102. doi:10.1046/j.1432-1033.2002.03020.x. PMID 12084049.
|
トピックス |
|
|
タイプ |
|
参考となる書籍
以下のリストは英語文献ばかりであるが、邦訳があるかもしれない。
入門書
- Athel Cornish-Bowden, Fundamentals of Enzyme Kinetics. (3rd edition), Portland Press 2004, ISBN 1-85578-158-1.
- Nicholas Price, Lewis Stevens, Fundamentals of Enzymology, Oxford University Press, 1999. ISBN 0-19-850229-X
- Tim Bugg, An Introduction to Enzyme and Coenzyme Chemistry Blackwell Publishing, 2004 ISBN 1-4051-1452-5
発展的な本
- Irwin H. Segel, Enzyme Kinetics : Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley-Interscience; New Ed edition 1993, ISBN 0-471-30309-7.
- Alan Fersht, Structure and Mechanism in Protein Science : A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, 1998. ISBN 0-7167-3268-8
- Santiago Schnell, Philip K. Maini, A century of enzyme kinetics: Reliability of the KM and vmax estimates, Comments on Theoretical Biology 8, 169–187, 2004 DOI: 10.1080/08948550390206768
- Chris Walsh, Enzymatic Reaction Mechanisms. W. H. Freeman and Company. 1979. ISBN 0-7167-0070-0
- Paul F. Cook and W.W. Cleland Enzyme kinetics and mechanism Garland Science, 2007 ISBN 0-8153-4140-7
外部リンク
全て英語である。
- Animation of an enzyme assay — 反応条件を変えたときの変化について
- MACiE — 酵素の反応機構データベース
- ENZYME — Expasy 酵素命名法データベース
- EzCatDB —触媒機構データベース
- BRENDA — 酵素データベースで、基質、阻害剤、反応式を掲載している。
- SABIO-RK - 反応速度論のデータベース
- Joseph Kraut's Research Group, University of California San Diego —いくつかの反応機構のアニメーション
- Symbolism and Terminology in Enzyme Kinetics —酵素速度論における概念と用語の解説
- An introduction to enzyme kinetics — 酵素速度論についてのオンラインチュートリアル
- Enzyme kinetics animated tutorial —音声つきのアニメーションによる酵素の解説
抗体酵素
抗体酵素(こうたいこうそ)とは、触媒活性を有するモノクローナル抗体のことである。抗体触媒、アブザイム(abzyme、antibody と enzyme の合成語)、あるいは catmab (catalytic monoclonal antibody から)ともいう。元来は人工的に創生されたものをいうが、ヒト体内にも見出されており、正常なヒトの抗-血管作動性小腸ペプチド(VIP:vasoactive intestinal peptide)抗体や、全身性エリテマトーデス(自己免疫疾患)患者の抗体(DNAに結合し加水分解する)がある。
酵素は反応過程で生じる(酵素がない場合には不安定な)中間体を安定化させることにより触媒機能を果たす。ある反応の中間体に類似した分子を結合するような抗体があれば、その抗体は中間体を安定化し、それによって反応を触媒できる可能性がある。このような戦略により、天然の酵素にないような酵素活性を有する抗体酵素を生み出す試みが行われている。抗体酵素はまたバイオテクノロジーにおいても、たとえばDNAに対して特異的な反応を起こすなど有用なツールとなる可能性がある。
外部リンク
抗体酵素:免疫システムを用いた新規生体触媒の創出
カテゴリ: 分子生物学生化学酵素抗体